



ISSN 1757-8515
Arrhythmogenic Right Ventricular Dysplasia Diagnosed Through Characterization of Cardiac Tissue in a Deceased Sibling: A Case Report
Raja Ezman Faridz Raja Shariff, Rizmy Najme Khir & Sazzli Kasim
Cite this article as: BJMP 2018;11(2):a1117
|
|
Abstract Introduction: Arrhythmogenic Right Ventricular Dysplasia (ARVD) is a rare cause of cardiomyopathy and sudden cardiac death. Often times, diagnosis relies on electrocardiography findings and magnetic resonance imaging of cardiac tissue, when available. Rarely, diagnosis is confirmed via histological evidence from an affected sibling. Case Report: We present a rare case of ARVD diagnosed via characterization of cardiac tissue of an affected, deceased sibling. A 21-year old gentleman presented to the emergency department following an episode of loss of consciousness. Chest radiography revealed cardiomegaly and electrocardiogram (ECG) revealed deep T-wave inversions in leads V2 to V4, with ventricular ectopic beats. Troponin-I levels were elevated at 480 pg/ml. It was revealed that the patient had a sibling who had died from an unknown cause, 5 years prior. His younger brother, 14 years of age at the time, had collapsed whilst playing basketball in a school compound. Unfortunately, he was pronounced dead on arrival to a medical facility. Autopsy findings revealed epicardial surfaces infiltrated with excessive fat tissue and with nodular fibrosis with cut sections showing diffuse transmural fibrofatty replacement of the right ventricular free wall extending to the endocardium involving right ventricular septum. This knowledge led to our patient having a cardiac MR performed, confirming a diagnosis of ARVD. Conclusion: The case highlights how having knowledge and confirmation of the inherited condition led to a quicker and more confident decision in managing a patient at high risk of SCD, as our patient was able to obtain an implantable cardiac defibrillator without much hesitation. Keywords: Case Report, Arrhythmogenic Right Ventricular Dysplasia, Arrhythmogenic Right Ventricular Cardiomyopathy, Cardiomyopathy, Heart FailureAbbreviations: ARVD: Arrhythromogenic Right Ventricular Dysplasia ECG: Electrocardiogram MRI: Magnetic Resonance Imaging ICD: Implantable Cardiac Defibrillator |
Background
Arrhythmogenic Right Ventricular Dysplasia (ARVD) was first described in a case series of 24 patients back in 1982 1, 2. Since then, our understanding of its pathophysiology has improved dramatically, with dedicated guidelines and literature being published to help with both diagnosis and management. Prompt diagnosis remains a struggle in majority of developing countries, including Malaysia, where resources and expertise are scarce, and obtaining both cardiac magnetic resonance imaging or endomyocardial biopsies remain a challenge. Furthermore, diagnosis is difficult in most cases as clinical presentation may vary and wide range of clinical mimics exist. We present a unique case of ARVD, diagnosed early through the knowledge of having a deceased sibling, whom had endomyocardial tissue characterization performed in the past confirming the presence of the disease in a first degree relative.
Case Report
A 21-year old gentleman presented to the emergency department following an episode of loss of consciousness, lasting approximately 30 minutes which recovered spontaneously. He denies having any similar episodes in the past. However, he had been suffering from reduced exercise tolerance, with a New York Heart Association (NYHA) Class II, over the past 1 year. He had no known medical illness at the time but smoked 6 cigarettes a day for the past 7 years.
His vital signs were stable on arrival, with a heart rate of 73 beats per minute, regular in rhythm, a blood pressure of 143/84 mmHg, respiratory rate of 19 breaths per minute, temperature of 37 degrees Celcius and oxygen saturation of 98% on room air. Cardio-respiratory examination revealed no murmurs, and normal heart and breath sounds. There were no carotid bruits audible. There was no evidence of any neurological deficits on neurological examination.
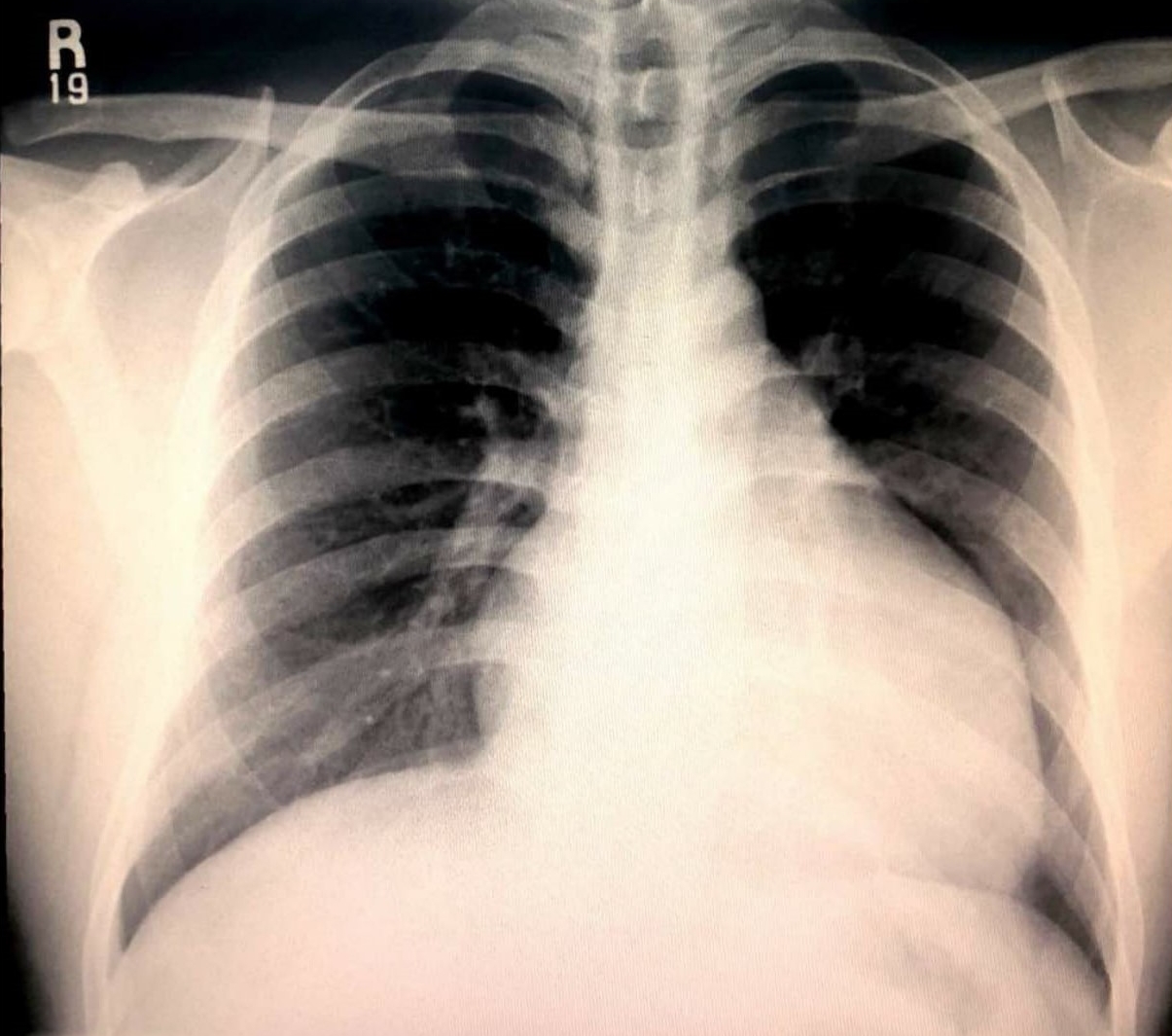
Figure 1 – Chest radiography demonstrative cardiomegaly
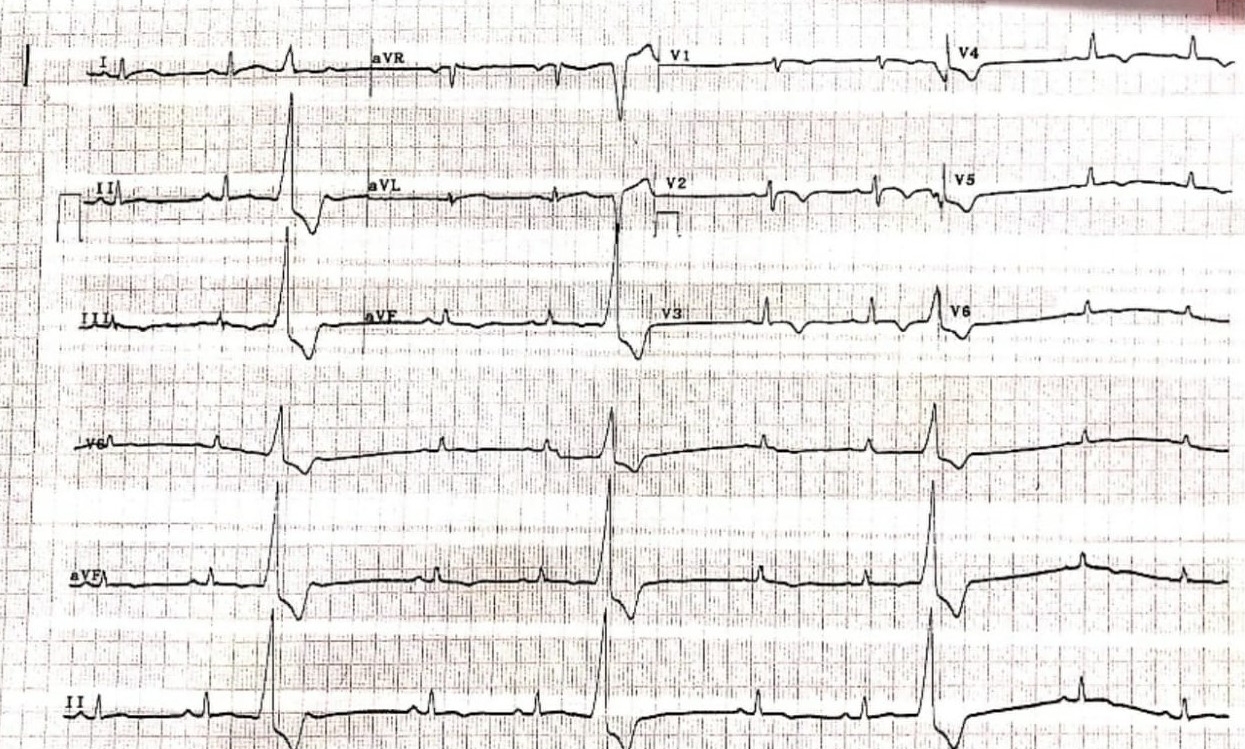
Figure 2 – Electrocardiogram revealing T-wave inversions in leads V2 to V4 with ventricular ectopic beats
Chest radiography revealed cardiomegaly (Figure 1). Electrocardiogram (ECG) revealed deep T-wave inversions in leads V2 to V4, with ventricular ectopic beats (Figure 2). Due to the suspicious-looking ECG, a serum Troponin-I test was performed, which was elevated at 480 pg/ml. The patient was treated for acute coronary syndrome complicated by cardiac syncope, and was later referred to the medical team for further inpatient management.
However, on further history, it was revealed that the patient had a sibling who had died from an unknown cause, 5 years prior. His younger brother, 14 years of age at the time, was brought in after collapsing whilst playing basketball in a school compound. Unfortunately, he was pronounced dead on arrival to the clinic. A post-mortem was performed due to the unexpected nature of the event. Fortunately, our patient was brought into the same hospital as his sibling, allowing us to trace previous autopsy reports and images, with consent.
Macroscopic examination of the right ventricular cavity revealed epicardial surfaces showing infiltration of excessive fat tissue with nodular fibrosis. The right ventricular cavity appeared dilated and cut sections showed diffuse transmural fibro-fatty replacement of the right ventricular free wall, extending into the endocardium and involving the right ventricular septum (Figure 3a).
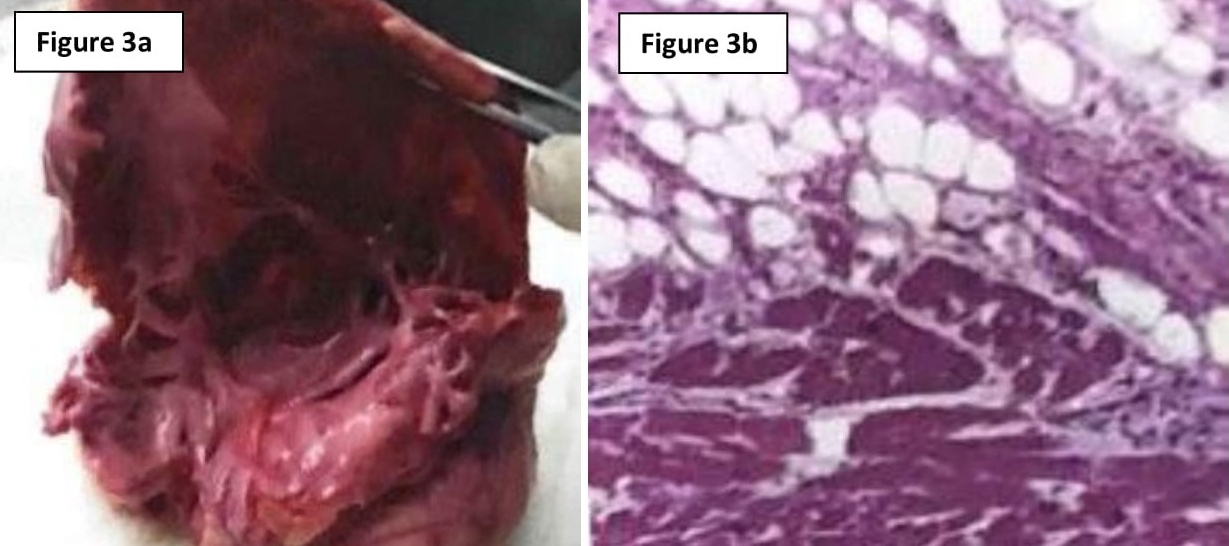
Figure 3a – Macroscopic examination of right ventricular cavity, which was dilated and showing signs of transmural fibrofatty infiltration. Figure 3b – Histological evidence of focal lymphocytic infiltration, myocyte hypertrophy and degenerative cytoplasmic changes.
Histology revealed extensive fatty infiltration with interstitial fibrosis, primarily in the epicardium. There was associated myocyte loss with hypertrophy of cardiac muscle cells remaining (Figure 3b). Both macroscopic and microscopic findings were suggestive of ARVD.
After learning of the autopsy results, changes in clinical management took place, with priorities being shifted towards obtaining an echocardiogram, cardiac Magnetic Resonance Imaging (MRI) and Holter recording, as opposed to diagnostic angiography and coronary evaluation. Echocardiography revealed an ejection fraction of 25 to 30%, with evidence of left ventricular dyssynchrony, a tethered posterior mitral valve leaflet with mild eccentric regurgitation, consistent with dilated cardiomyopathy.
Cardiac MRI revealed both left and right ventricular dilatation, end diastolic dimensions being 5.8 cm and 4.4 cm and end-diastolic volume being 153 ml/m2 and 149 ml/m2 respectively, with evidence of bi-ventricular dyssynchrony. Left ventricular and right ventricular ejection fraction measured 31% and 8% respectively. There was also bilateral atrial dilatation. Gadolinium study revealed late enhancement in areas of the right ventricular wall (Figure 4).
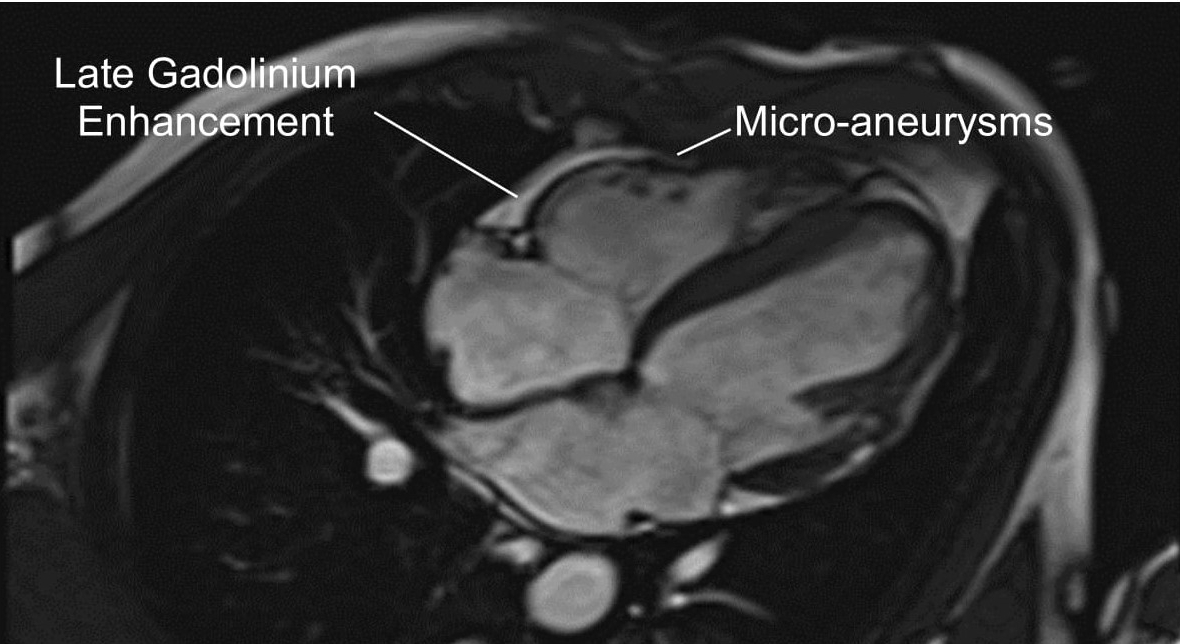
Figure 4 – Four-chamber view of cardiac magnetic resonance imaging revealing evidence of right ventricular enhancement following gadolinium study.
A 24-hours Holter recording revealed up to significant ventricular ectopic burden, many of which were bigeminy and trigeminy in nature. In view of symptoms and the above investigative findings, the patient consented to insertion of an implantable cardiac defibrillator (ICD) 4 weeks later in our centre, and has since recovered well with regular monitoring.
Discussion
ARVD is rare, prevalence ranging between 1 in 2000 to 1 in 5000 (taking into consideration geographical variations) and accounts for 5% of deaths in young adults and 25% of deaths in athletes 3, 4. Typical histopathological feature of ARVD is the loss of right ventricular myocardium, replaced heavily by fibro-fatty tissue. Less commonly, left ventricle involvement have been reported 5, 6. Consequence from such pathological process leads to arrhythmias, heart failure and more importantly sudden cardiac death (SCD), with mortality rate ranging between 4 to 20% and peaking in the fourth decade, equally in both males and females 5.
Diagnosis is difficult in most cases as clinical presentation may vary and wide range of clinical mimic exist, including myocarditis, sarcoidosis, Brugada syndrome, idiopathic RV outflow tract VT and congenital heart diseases with right chambers overload amongst others 6. A Diagnostic Criteria was developed in 1994, with further modification in 2010 to assist in the diagnosis of ARVD and although the criterion has been shown to be specific, it lacks sensitivity 7. Nevertheless, it highlights several key areas, a mix of clinical, radiological, histological and electrophysiological features, that assist in diagnosis 4.
Despite not having any further evaluation or investigations performed at the time of presentation, in view of circumstances, our patient’s deceased sibling had supportive histological features. Therefore, our patient met the major criteria of having a first degree relative affected by the disease. More importantly, the suspicious family history had prompted further evaluation for the disease, allowing the medical team to prioritize investigations performed, specifically the Cardiac MRI and Holter evaluation. This led to early risk stratification and decision to implant an ICD for the patient, as he was deemed at high risk of SCD.
Conclusion
This case highlights the importance of good history taking, including a detailed family history of SCD or cardiac-related diseases, especially in young patients presenting with typical cardiac-related symptoms. Early identification and appreciation of risk will subsequently affect the outcomes of such patients affected by ARVD. Furthermore, important diagnosis like ARVD will have implications to relatives and future off-springs, further highlighting the need for detailed evaluation of patients similar to ours described in the above case report.
|
Competing Interests None declared Author Details RAJA EZMAN FARIDZ RAJA SHARIFF (MBCHB, MRCP), Universiti Teknologi Mara, Sungai Buloh, Malaysia. RIZMY NAJME KHIR (MBBCH, MRCP), Universiti Teknologi Mara, Sungai Buloh, Malaysia. SAZZLI KASIM (MBBCh, BAO, BMedSc, MRCPI, CSCST, FNHAM), Universiti Teknologi Mara, Sungai Buloh, Malaysia. CORRESPONDENCE: RAJA EZMAN FARIDZ RAJA SHARIFF, Universiti Teknologi Mara Sungai Buloh Campus, Jalan Hospital, Sungai Buloh, 47000, Selangor, Malaysia. Email: rajaezman@gmail.com |
References
- Marcus FI, Fontaine GH, Guiraudon G, et al. Right Ventricular Dysplasia: A Report of 24 Adult Cases. Circulation. 1982; 65:384–98.
- Sosman MC. ‘Illustrative Echocardiographic Cases’ in The Disorders of Cardiac Rhythm. Ed: Schamroth L. Blackwell. Oxford and Edinburgh. 1971; 335.
- Basso C, Pilichou K, Bauce B, et al. Diagnostic Criteria, Genetics, and Molecular Basis of Arrhythmogenic Cardiomyopathy. Heart Failure Clin. 2018; 14:201–213.
- Rao U, Agarwal S & Gilbert TJ. Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC): Case Report and Review of Literature. Heart Asia. 2014; 6:145–149.
- Hoorntje ET, Rijdt WP, James CA, et al. Arrhythmogenic Cardiomyopathy: Pathology, Genetics, and Concepts in Pathogenesis. Cardiovascular Research. 2017; 113:1521–1531.
- Corrado D, Link MS & Calkins H. Arrhythmogenic Right Ventricular Cardiomyopathy. N Engl J Med. 2017; 376:61-72.
- Wang W, James CA, & Calkins H. Diagnostic and Therapeutic Strategies for Arrhythmogenic Right Ventricular Dysplasia / Cardiomyopathy Patient. Europace. 2018; 0:1–13.

The above article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.