Although it is well appreciated that pancreatitis is frequently secondary to biliary tract disease and alcohol abuse, it can also be caused by drugs, trauma and viral infections, or even be associated with metabolic and connective tissue disorders.1 Knowledge of the true incidence of drug-induced acute pancreatitis is dependent on the clinician’s ability to exclude other possible causes, and by promptly reporting the occurrence. Based on individual case reports and case control studies of drug-induced acute pancreatitis, the estimated overall incidence ranges from between 0.1 and 2% of pancreatitis cases.2,3 In particular, drug-induced acute pancreatitis is of mild severity and usually resolves without significant complications.4
Attempts have been made to categorize the risk of drugs causing acute pancreatitis. A previous classification system described by Mallory and Kern Jr. categorized drugs associated with acute pancreatitis as definite, probable, or possible.5 Trivedi et al. proposed a newer classification system for commonly used medications associated with drug-induced pancreatitis. Class I drugs are those medications with at least 20 reported cases of acute pancreatitis and at least one case with a positive rechallenge. Drugs with fewer than 20 but more than 10 reported cases of acute pancreatitis, with or without a positive rechallenge, are designated into Class II. While those medications with 10 or less reported cases, or unpublished reports in pharmaceutical or FDA files, are grouped into Class III.6
Acute pancreatitis as a result of either doxorubicin or cyclophosphamide, or a combination of both, or fluorouracil or epirubicin is a rare occurrence and has seldom been reported in the literature. Even the drug package labels registered with the FDA do not indicate the possible occurrence of pancreatitis. In this case report, we present a rare occurrence of drug-induced acute pancreatitis after the completion of the first cycle of the chemotherapy protocol involving cyclophosphamide and doxorubicin in a patient with stage 3 breast cancer, with recurrences of acute pancreatitis after re-challenging with cyclophosphamide and a derivative of doxorubicin, given individually on two separate occasions.
CASE PRESENTATION
A 58 year-old female presented to the emergency room with a one day history of severe, diffuse, deep-seated abdominal pain that radiated to her back, associated with nausea and vomiting, and was unrelieved despite the intake of NSAIDs. There was no reported fever, chills, diarrhea, dysuria, or antecedent trauma. Her medical history is notable for well-controlled hypertension, hyperlipidemia and hypothyroidism for which she takes amlodipine, atorvastatin and levothyroxine. She was recently diagnosed with left-sided breast cancer, Stage III, two months prior to admission and underwent a left modified radical mastectomy. Three days prior to the hospital visit, she was given her first cycle of chemotherapy with Doxorubicin 60 mg/m2 and Cyclophosphamide 600 mg/m2 along with Pegfilgrastim 6 mg and Fosaprepitant 150 mg. She is a former cigarette smoker, drinks alcohol infrequently, and denies illicit drug use. Her family history is unremarkable.
Physical examination revealed stable vital signs without a fever (36.6◦C). She had non-icteric sclerae and a dry oral mucosa. Chest exam revealed a well-healed left mastectomy scar and an infusaport located on the right anterior chest wall. Her breath sounds were clear bilaterally. Her heart sounds were normal. Her abdominal exam was significant for mild tenderness to palpation in the epigastric area without palpable masses, organomegaly or ascites. There was no evident ecchymosis observed. The extremities were warm to touch with intact and symmetrical pulses, and without bipedal edema.
Initial work-up revealed an elevated leukocyte count of 42,000 with 80% neutrophils and 17% band forms. Basic metabolic panel was normal except for mild hyponatremia of 129 mEq/L. Serum amylase and lipase were markedly elevated at 2802 units/L and >2000 units/L, respectively. Liver function panel was normal (Alk phos 63 U/L [ref range 30-115 U/L], GGT 21 U/L [ref range 3-40 U/L], total bilirubin 0.90 mg/dL [ref range 0-1.5 mg/dL]). The coagulation profile was within normal range. Imaging of the abdomen with a CAT scan with intravenous and oral contrast showed haziness in the pancreatic fat plane suggestive of pancreatic inflammation, with no gallstones, focal abscesses, hepatic masses, or biliary ductal dilatation (Figure 1). Right upper quadrant ultrasound was essentially normal (Figure 2).
Figure 1. Coronal view of CT scan of abdomen and pelvis with IV and oral contrast showing haziness in the peripancreatic fat plane.
Figure 2. Sonogram of the right upper quadrant of the abdomen showing gallbladder devoid of gallstones and non-dilated common bile duct.
She was admitted to the medicine unit with the assessment of acute pancreatitis likely secondary to doxorubicin and cyclophosphamide. Intravenous fluid hydration with normal saline was initiated. She was kept NPO (nothing per os) and was started on empiric Imipenem and IV Esomeprazole. Her abdominal pain was controlled with intravenous morphine and her nausea with Ondansetron as needed. The serial basic chemistry panel was monitored and electrolyte deficits were replaced accordingly. Further work-up was performed to identify other possible etiologies of pancreatitis. The lipid panel was within normal limits (cholesterol 169 mg/dL [0-200 mg/dL], HDL 74 mg/dL, LDL 71 mg/dL and triglycerides 54 mg/dL [0-150 mg/dL]). The serum calcium levels remained within the normal range throughout her hospital stay. An abdominal sonogram demonstrated absence of gallstones or dilatation of the common bile duct, with a normal appearing liver parenchyma and pancreas. During her stay in the medicine unit, the patient’s abdominal pain improved and she was gradually started on an oral diet, which she tolerated well. Her serum electrolytes remained stable, while her serial CBC revealed progressively decreasing trends in leukocytes, hemoglobin, hematocrit, and platelet count, findings which were attributed to her prior chemotherapy. Repeat serum amylase and lipase both trended downward. The patient was discharged with follow up in the Oncology clinic. A month later, she was started on another chemotherapy regimen that consisted of weekly administration of Paclitaxel 80 mg/m2 which, over the next two months, the patient completed without any complications. Then, after explaining the risks of recurrent pancreatitis, the patient consented to have a trial of cyclophosphamide 500 mg/m2 along with fluorouracil 500 mg/m2. Five days after receiving the chemotherapy, the patient developed acute pancreatitis which was attributed to cyclophosphamide. She again made a full recovery at that time. Three weeks later, her chemotherapy regimen was again changed, to epirubicin 90 mg/m2 and fluorouracil. Four days after receiving this regimen, she again, for the third time, had a recurrence of acute pancreatitis. At this time, a repeat abdominal sonogram revealed a 4mm echogenic focus adherent to the anterior gallbladder wall with a comet tail sign, suggestive of cholesterol crystals lodged within Rokitansky-Aschoff sinuses of the gallbladder wall. There were no visible gallstones. A subsequent MRI of the abdomen with contrast revealed a small rounded hypointensity in the dependent portion of the gallbladder wall that was suggestive of a gallstone, however, there was no biliary obstruction, choledocholithiasis or an obstructing pancreatic mass. At this point, chemotherapy was stopped and anastrozole along with radiation therapy was initiated. The patient continues to be followed regularly and has had no recurrence of pancreatitis since her last episode.
DISCUSSION
The case presented described the development of acute pancreatitis in a patient with breast cancer three days after receiving the chemotherapy regimen consisting of cyclophosphamide and doxorubicin. After re-challenging the patient with cyclophosphamide, and again a few weeks later with a derivative of doxorubicin, epirubicin, acute pancreatitis recurred on each occasion. Despite the presence of cholelithiasis detected on the abdominal MRI, the temporal presentation of acute pancreatitis after chemotherapy exposure is highly suggestive of the role these chemotherapeutic agents played in triggering these three acute attacks. Acute pancreatitis was diagnosed based on the clinical suspicion and symptoms suggestive of the acute pancreatitis and was supported by the marked elevation in serum amylase and lipase, as well as, the radiologic evidence of pancreatic inflammation, both of which are markers of acute pancreatitis.
Chemotherapy-induced acute pancreatitis involving cyclophosphamide and doxorubicin either alone or in combination, is quite rare that even the drug labels registered with the FDA do not indicate acute pancreatitis as one of the possible complications. This scenario highlights the importance of drug surveillance and prompt reporting in order to maintain a credible drug safety database.
Though the drug latency, which is the interval between the initial exposure to the drug and the development of acute pancreatitis, differs variably, the present case is considered to have an intermediate latency (1-30 days). Other drugs may have short (< 24 hours) or long (>30 days) latency periods. Examples of drugs with short latency are acetaminophen, codeine, erythromycin and propofol. Intermediate latency drugs include L-asparaginase, pentamidine and stibugluconate. Drugs with long latency are estrogen, tamoxifen, valproate and dideoxyinosine.7
Based on the revised classification of Badalov et al, the combination of cyclophosphamide and doxorubicin is classified as Class IV drugs, which have the weakest association with acute pancreatitis due to limited information and the lack of adequate detailed case reports. Fluorouracil, which has been known to cause a gastrointestinal ulcer, is also categorized as a Class IV drug, while epirubicin, which is derived from doxorubicin, has not been classified, as it has not been reported before to cause acute pancreatitis. In implicating drugs in the etiology of acute pancreatitis, two conditions must be considered to weigh the strength of the association between the causality and the disease process, namely: a positive rechallenge test resulting in the recurrence of pancreatitis and a similar latency period between the drug exposure and development of the disease.7
The combination of drugs rather than a single agent was implicated for drug-induced pancreatitis in a previous case report that described the development of acute pancreatitis shortly after the second cycle of the chemotherapy regimen composed of cyclophosphamide, doxorubicin, and vincristine in a patient with mediastinal immunoblastic lymphoma. The pancreatitis episode resolved over 48 hours without complications.8
Another case was described in a patient with breast cancer developing acute pancreatitis four days after the third cycle of chemotherapy, which involved docetaxel and carboplatin.9
Toprak et al. reported the occurrence of acute pancreatitis in a patient with multiple myeloma after the initial treatment with the triple regimen chemotherapy protocol consisting of vincristine, doxorubicin, and dexamethasone. In this case report, symptoms suggestive of acute pancreatitis started to manifest on the first day of the treatment, with resolution following discontinuation of the drugs.10
Other antineoplastic agents for breast cancer associated with drug-induced pancreatitis are alemtuzumab, trastuzumab and tamoxifen. Extended use of these medications may cause chronic pancreatitis as a result of their causing repeated clinical or subclinical episodes of acute pancreatitis.6 Most cases of drug-induced pancreatitis follow a mild clinical course.7
In a retrospective study involving 1613 patients diagnosed with acute pancreatitis in a gastroenterology center, the incidence of drug-induced pancreatitis had been reported in 1.4% of patients treated for acute pancreatitis. It has been observed that a higher incidence of drug-induced acute pancreatitis occurs in elderly or pediatric patients, and in those patients with inflammatory bowel disease or AIDS.11
The pathophysiology behind drug-induced pancreatic injury remains unclear. Potential mechanisms underlying such pancreatic injury might be related to drug hepatotoxicity which can be secondary to intrinsic toxicity of the drugs affecting the tissue, or due to an idiosyncratic reaction. In the vast majority of the cases, an idiosyncratic reaction could be the main pathway for tissue injury through a hypersensitivity reaction or production of toxic intermediate metabolites. Idiosyncratic reactions have a longer latency period of months to years before the onset of pancreatitis while the onset of hypersensitivity reactions is earlier (i.e. 1-6 weeks).7
CONCLUSION
Due to a variable latent period between the initial drug exposure and the onset of clinical symptoms, drug-induced pancreatitis must remain as a differential diagnosis in patients receiving chemotherapy regimens and presenting with the constellation of symptoms typical of acute pancreatitis. Due to the unclear pathogenesis of chemotherapy-induced pancreatitis, post-marketing surveillance and adverse drug reporting are paramount in elucidating the effect these drugs have on the pancreas.
Various classes of medications have been known to cause drug induced liver injury (DILI), however not much literature has been published regarding angiotensin converting enzyme inhibitors (ACE-I) causing DILI. Recent years have seen tremendous increases in ACE-I prescriptions for coronary artery disease, diabetic nephropathy and hypertension. We report the first case of lisinopril induced hepatitis via a cholestatic mechanism.
Case:
A 47 year old female with history of diabetes mellitus type 2, hypertension, chronic kidney disease (CKD)stage III, non-obstructive coronary artery disease was admitted with complains of generalized weakness, lack of appetite, yellow discoloration of skin and eyes, dark urine and white stools for 1 week prior to admission. She denied history of alcohol abuse, past liver disease, illicit drug use, recent sick contacts, fever, chills, travel. Current patient medications included lisinopril, pioglitazone, furosemide, atenolol, metformin and detemir. Patient was started on these medications about 2 years prior to admission. Patient received enalapril for 5 months before switching to lisinopril about 2 years prior to presentation.
Physical examination was positive for icteric sclera, icteric skin; negative for spider nevi, palmar erythema and asterixis. Exam did not reveal hepatomegaly or splenomegaly. Labs showed hemoglobin 8.7 gm/dl, normal white count and platelet, normal C-reactive protein, alkaline phosphatase (ALP) 750 U/L, aspartate transaminase (AST) 169 U/L, alanine transaminase (ALT) 210 U/L, gamma-glutamyl transferase (GGT) 813 U/L, total bilirubin 13.4mg/dl with conjugated fraction 7.7mg/dl, ammonia level 64. Prior to initiation of lisinopril ALP was 87 U/L, GGT 53 U/L, with AST18 U/L, ALT 11 U/L and normal bilirubin fractions. Hepatitis A, B, C and D serologies were negative. Serum acetaminophen level was normal. Anti nuclear antibody (ANA), anti- mitochondrial antibody (AMA), anti-endomysial antibody, c-anti-neutrophil cytoplasmic antibody (ANCA), p-ANCA was negative. Anti smooth muscle antibody was weakly positive in titre of 1: 40. Creatine kinase, ceruloplasmin and alpha -1 antitrypsin level were normal. Quantiferon gold was negative. Lipid panel was deranged with cholesterol level 1017 and low density lipoprotein 1006, triglycerides 255.
Ultrasonography and magnetic resonance imaging abdomen showed hepatomegaly 17.5cms but was negative for fatty infiltration of liver, stones, cirrhotic features or dilation of biliary tree. Liver biopsy was done which showed mild portal chronic hepatitis with lymphocytic infiltration (Fig: 1), cholestasis (Fig: 2), mild portal fibrosis (Fig: 3), negative for bile duct damage (Fig: 4), negative for cytoplasmic inclusion. Congo red stain was negative for amyloid.
Figure 1: Mild hepatitis with portal tract lymphocytic infiltration.
Patient was treated with fluids, anti-histaminic, ursodeoxycholic acid. Patient was unable to tolerate coleveselam. Impression was drug induced hepatitis, lisinopril was discontinued and patient improved clinically and biochemically. Discharge labs two weeks after discontinuation of lisinopril showed AST 80 U/L, ALT 70 U/L, ALP 1045 U/L and GGT 1212 U/L; total bilirubin of 3.93 mg/dl with conjugated fraction 2.43mg/dl. Patient was discharged uneventfully with follow up in Hepatology clinic. Six months after discontinuation of lisinopril ALP was 199 U/L, GGT 168 U/L with AST 19 U/L, ALT 17 U/L, total bilirubin 0.9mg/dl and conjugated bilirubin 0.21mg/dl. Patient is currently asymptomatic and icterus has resolved.
Discussion:
ACE-I has been used widely for coronary artery disease, hypertension and diabetic nephropathy and approximately 159 million prescriptions for ACE-I are written annually. Recent JNCC guidelines recommended ACE-I to be used as first line anti-hypertensives for patients with CKD and diabetes. The common side effects known about ACE-I use are cough and angioedema, hypersensitivity. However not much awareness exists regarding ACE-I induced hepatotoxicity. It is important to consider ACE-I as an etiology for drug-induced liver injury (DILI) since continuation of the ACE-I beyond onset of hepatitis is fatal1.
Literature review shows multiple reports of DILI with captopril2, 3, ramipril4, fosinopril5, 6 and enalapril.2,7 Most commonly implicated ACE-I are enalapril and captopril. The usual presentation for ACE-I induced hepatotoxicity is cholestasis mediated hepatitis. Till date there have been four case reports published reporting lisinopril as cause of hepatitis 1, 8, 9 All 4 cases of lisinopril induced hepatotoxicity have shown a hepatocellular pattern of liver injury and did not show any cholestatic features. We report the first case of lisinopril induced cholestasis mediated hepatotoxicity.
In our case, patient had received enalapril for 5 months before initiation of lisinopril; however patient developed symptoms 2 years after initiation of lisinopril. The patient had no past medical history of liver or biliary tract disease. A thorough investigative workup was negative for autoimmune and other viral causes of hepatitis. Older case reports of lisinopril induced toxicity have shown similar histopathological findings of portal inflammation by lymphocytes without centrilobular zonal necrosis.9 There are various theories regarding possible mechanisms for DILI with lisinopril, namely terminal proline ring mediated bile stasis8, 10 and hypersensitivity to the sulfhydryl group.2 Discontinuation of metformin, pioglitazone, furosemide, atenolol and detemir did not result in clinical or biochemical improvement. Patient was initially continued on lisinopril since suspicion was low and then later discontinued. Similarity in histopathological findings along with a strong temporal relationship between lisinopril withdrawal and improved biochemical and clinical scenario, with absence of other constitutional symptoms and eosinophilia strongly point toward lisinopril-induced hepatotoxicity.
Our case had a long period of latency between drug intake and onset of hepatic injury which is consistent with other published reports of lisinopril induced hepatocellular injury9, 10, 11; however the mechanism responsible for latency or hepatotoxicity remains unclear. Earlier report postulate metabolic idiosyncratic reaction as a possible molecular mechanism for hepatocellular injury9. However our case is unique as the primary mode of injury appears to be cholestatic. Since our patient received enalapril before initiation of lisinopril without any adverse events, this case adds further controversy as to whether this patient could have been safely continued on other ACE-I except lisinopril or whether she would have developed hepatotoxicity if enalapril was continued. This case highlights further need for research to evaluate ACE-I induced hepatotoxicity. Currently the awareness for ACE-I induced liver injury is low and there are no guidelines guiding physician to monitor for possible hepatic adverse events. Further research is needed to delineate the mechanism by which ACE-I cause hepatotoxicity and to define possible risk factors.
Conclusion:
Discontinuation of ACE-I beyond recognition of DILI hepatitis usually leads to normalization of liver enzymes, however continuing or reinitiating ACE-I can be severe and potentially fatal. Thus, it is important to be aware of ACE-I as a possible cause of DILI, which can present with either hepatocellular or cholestatic mechanism and to promptly discontinue ACE inhibitor use. Currently there are no guidelines in place for monitoring of liver enzymes following initiation of ACE-I and more research is required to delineate possible mechanisms and prevent further DILI in such patients.
Community acquired urinary tract infection (UTI) due to Escherichia coli is one of the most common form of bacterial infections, affecting people of all ages. Originally ESBL (extended spectrum β-lactamases) producing E. coli was isolated from hospital setting but lately this organism has begun to disseminate in the community.1
In India community presence of ESBL producing organisms has been well documented. However, various epidemiological factors associated with ESBL producing strains need to be documented. This will allow clinicians to separate patients with community UTI with these factors so that appropriate and timely treatment can be given.2 A community UTI when complicated may be a potentially life-threatening condition. In addition, for deciding the empirical treatment for patients with a UTI a thorough knowledge of local epidemiology is required. Therefore, the primary objective of this study was to determine the epidemiological factors associated with ESBL positive community acquired uropathogenic E. coli isolates and to determine their susceptibility to newer oral drugs. Mecillinam is a novel β-lactam antibiotic that is active against many members of family Enterobacteriaceae. It binds to penicillin binding protein (PBP 2), an enzyme critical for the establishment and maintenance of bacillary cell shape. It is given as a prodrug that is hydrolyzed into active agent. It is well tolerated orally in the treatment of acute cystitis.3
Material and Methods
This prospective study was conducted, from Jan 2012- July 2012, in our tertiary care hospital, which caters to medical needs of the community in North India.
Study Group:
The study group included patients diagnosed as having a UTI in outpatient clinic, or the emergency room or patients diagnosed within 48 hrs after of hospitalization. These patients and were labeled as patients having a community UTI. A diagnosis of symptomatic UTI was made when patient had at least one of the following signs or symptoms with no other recognized cause: fever ≥ 38.8˚C, urgency, frequency, dysuria or suprapubic tenderness and a positive urine culture (i.e. ≥105 microorganisms/ml of urine).4 Various epidemiological factors for each patient were recorded on individual forms. This included age, presence of diabetes mellitus, renal calculi, pregnancy, history of urinary instrumentation, recurrent UTI (more than 3 UTI episodes in the preceding year) and antibiotics intake (use of β-lactam in the preceding 3 months).2
Patients with a history of previous or recent hospitalization were excluded from study.
Antibiotic susceptibility testing was carried out following Clinical Laboratory Standards Institute (CLSI) guidelines using the Kirby-Bauer disc diffusion method.5 The antibiotics, which were tested included Amoxyclav (30/10µg), Norfloxacin (10µg), Ciprofloxacin (5µg), Tetracycline (30µg), Nitrofurantoin (300µg), Trimethoprim-sulfamethoxazole (23.75/1.25µg), Cephalexin (30µg), Cefaclor (30µg), Cefuroxime (30µg), Mecillinam (10µg) (Hi-Media, Mumbai, India).
Detection of ESBL
ESBL detection was done for all isolates according to latest CLSI criteria.5
Screening test - According to latest CLSI guidelines, zone diameter of E. coli strain for Ceftazidime <22mm and for Cefotaxime < 21mm is presumptively taken to indicate ESBL production.
Confirmatory test - As per CLSI guidelines, ESBLs were confirmed by placing a disc of Cefotaxime and Ceftazidime at a distance of 20mm from a disc of Cefotaxime /Clavulanic acid (30/10µg) and Ceftazidime/Clavulanic acid (30/10µg) respectively on a lawn culture of test strain (0.5 McFarland inoculum size) on Mueller-Hinton agar. After overnight incubation at 37° C, ESBL production was confirmed if there was a ≥5mm increase in zone diameter for either antimicrobial agent tested in combination with Clavulanic acid versus its zone when tested alone
Control strain - Standard strain of Klebsiella pneumonia ATCC 700603 was used as ESBL positive controland Escherichia coli ATCC 25922 was used as ESBL negative control.
Results
Out of total of 140 strains of E. coli, which were screened for ESBL production, 30 (21.4 %) isolates were found to be positive. High-level resistance was seen for many antimicrobial agents like Cephalexin (92.8%), Cefaclor (90%), Amoxy-clavulanate (88.57%), Cefuroxime (75.7%), Sulfamethoxazole-trimethoprim (72.8%), Norfloxacin (75.71%) and Ciprofloxacin (70%). Sensitivity to Nitrofurantoin was found to be 90%. Only 4.5% of uropathogenic E. coli were resistant to Mecillinam.
Various epidemiological factors seen in ESBL producers include female patients (n =24, 80%), history of antimicrobial intake (n = 17,57 %), elderly age >60 years (n =16 53%), renal calculi (n =15, 50%), history of recurrent UTI (n =11, 37 %), pregnancy (n = 11,37%), diabetes mellitus (n = 7, 23%) and history of urogenital instrumentation (n = 7, 23%).
Discussion
The epidemiology of ESBL positive uropathogenic E. coli is becoming more multifaceted, with increasingly indistinct boundaries between the community and hospital.6 In addition, infection with an ESBL producing organisms causing community UTI is associated with treatment failure, delayed clinical response, higher morbidity and mortality. These organisms are multi-resistant to other antimicrobials like Aminoglycosides, Quinolones and Co-trimoxazole. Therefore, empirical therapy with Cephalosporins and Fluoroquinolones often fail in patients with community UTI.7
The rate of ESBL producers in our study is lower than that described by other authors. In a similar study Mahesh E et al. reported higher rate (56.2%) of ESBL positivity from E. coli, which were causing UTIs from a community setting.8 Additionally Taneja N et al. described a higher rate (36.5%) of ESBL positivity in uropathogens. 9,10
A high rate of resistance was seen to almost all antimicrobial agents. This is in agreement with other authors like Mahesh et al. and Mandal J et al.8,11 Mecillinam showed very good results with only 4.5% resistance. Wootton M et al. reported similar high activity of Mecillinam against E. coli(93.5%).3 Auer S et al. reported that Mecillinam can be a good oral treatment options in patients with infections due to ESBL organisms.7
A limitation of our study was that being a developing country with limited resources, molecular typing and determination of antimicrobial resistance profiles of the isolates was not done. In our study female patients, elderly, patients with a history of antimicrobial intake, renal calculi and history of recurrent UTI were important factors for infection due to ESBL producers. These findings are similar to risk factors studied by other authors.2 In conclusion; this study confirms that ESBL-producing E. coli strains are a notable cause of community onset infections especially in predisposed patients. The widespread and rapid dissemination of ESBL-producing E. coli seems to be an emerging issue worldwide. Further clinical studies are needed to guide clinicians in the management of community onset infections caused by E. coli.
A 40 year old patient presented to the hospital outpatient department with one year history of cough, choking sensation following swallowing, hoarseness of voice and loss of weight. History revealed his previous hospital admission 1 year back for management of organophosphorus poisoning during which he was intubated and put on mechanical ventilator for 10 days. Patient developed the symptoms a month after his discharge from the hospital. Cranial nerve examination was within normal limits. What is the possible diagnosis?
Gastro-oesophageal reflux disease
Tracheo-oesophageal fistula
Oesophageal diverticula
Oesophageal rupture
Fig 1: Barium swallow illustrating a dilated oesophagus and the TOF with resultant contamination of the trachea and bronchial tree
Fig 2: Oesophagoscopy showing TOF
Correct answer:
2. Tracheo-oesophageal fistula
Discussion:
A tracheo-oesophageal fistula (TOF) is a communication between the trachea and oesophagus which can be congenital or acquired. Congenital and acquired TOFs are associated with multiple complications, including poor nutrition, recurrent pneumonia, acute lung injury, acute respiratory distress syndrome, lung abscess, bronchiectasis from recurrent aspiration, respiratory failure, and death. Acquired TOFs occur secondary to malignant disease, infection, ruptured diverticula, and trauma.1, 2 Acquired TOFs are quite rare, and incidence rates have not been well documented. Post intubation TOFs uncommonly occur following prolonged mechanical ventilation with an endotracheal or tracheostomy tube. TOFs caused by endotracheal tube intubation depend on several factors, including prolonged intubation, an irritating or abrasive tube, and pressure exerted by the cuff. Pressures exceeding 30 mm Hg can significantly reduce mucosal capillary circulation and result in tracheal necrosis. Cuff pressure is particularly risky when exerted posteriorly against a rigid nasogastric tube in the oesophagus. Poor nutrition, infection, and steroid use cause tissue alteration, which predisposes patients for the development of TOFs. As a result of laryngeal bypass, spillage of oesophageal contents occurs into the trachea. Saliva, food and gastric juice contaminate the airways. This leads to congestion, infection, pneumonia, bronchial obstruction, atelectasis and respiratory distress. The severity of contamination depends on the width and length of the fistula as well as the posture of the patient. Spontaneous closure of non-malignant TOFs is exceptional.
Patients with acquired TOFs have high mortality and morbidity rates because of critical illnesses and co-morbidities. Acquired TOFs may occur in individuals of any age, and elderly individuals are at increased risk if they become ventilator dependent because of respiratory failure. Acquired TOFs can be diagnosed by instillation of contrast media into the oesophagus (Fig.1) or during direct visualization by flexible oesophagoscopy (Fig.2) or bronchoscopy. A high index of suspicion is needed to diagnose tracheo-oesophageal fistula in a post intubated patient presenting with symptom of cough following deglutition. Since acquired TOFs do not close spontaneously, surgical repair is needed if the patient is stable enough.3, 4 Critically ill patients are managed conservatively until stable enough for a major surgical procedure.
Typical oesophageal symptoms of gastro-oesophageal reflux disease include heartburn, regurgitation and dysphagia. The classic presentation of spontaneous oesophageal rupture is chest pain and subcutaneous emphysema after recent vomiting or retching (Mackler’s triad) in a middle-aged man with a history of dietary over-indulgence and over consumption of alcohol. Oesophageal diverticula presents with oropharyngeal dysphagia, usually to both solids and liquids, which is the most common symptom. Retention of food material and secretions in the diverticulum, particularly when it is large, can result in regurgitation of undigested food, halitosis, cough, and even aspiration pneumonia. The patient may note food on the pillow upon waking up in the morning.
An 87-year-old gentleman was admitted after sudden dysarthria and left facial palsy due to a right internal carotid artery occlusion. On examination, incidental spontaneous movements were seen at rest in the left leg (video), with bilaterally diminished Achilles reflexes. Patient was unaware of this finding. Muscle atrophy and hypoesthesia were not present. When walking on heels, left foot dorsiflexion was impaired.
What kind of physical finding is shown in this video?
A. Myoclonus B. Dystonia C. Tremor D. Chorea E. Fasciculation F. Myokymia
Answer / Discussion
Focal fasciculations in the tibialis anterior muscle are shown. When walking on heels, left foot dorsiflexion was slightly impaired.
Fasciculation is a brief, twitching, spontaneous involuntary contraction affecting muscle fibres served by one motor unit, which may be visible under skin. When present, they reflect denervation.
A complete history intake and neurological examination will lead to a sensible diagnostic work-up and to set a prognosis. Clinical differential diagnosis is presented in table 1.
Table 1: Key points for clinical diagnosis
Myoclonus
Brief, shocklike involuntary contraction of a muscle or group of muscles
Dystonia
Involuntary muscle contraction that can cause slow repetitive movements or abnormal postures
Tremor
Involuntary rhytmic contraction of antagonistic muscles
Chorea
Involuntary irregular movement that starts in one part of the body and moves unpredictably and continously to another part, like “dancing”
Myokymia
Involuntary spontaneous quivering, writhing movements within a single muscle not extensive enough to cause a movement of a joint
Localization helps in diagnosis: fasciculations can be generalised, in metabolic-toxic conditions, the benign fasciculation syndrome and degenerative disorders of anterior horn of spinal cord, as amyotrophic lateral sclerosis; segmental, as in syringomyelia; or focal, affecting the muscles controlled by a nerve or spinal root. When fasciculations are in a distribution that cannot be explained by plexus, root or nerve lesion amyotrophic lateral sclerosis (ALS) must be ruled out as soon as possible.
Evolution findings are also pivotal. The absence of muscle atrophy suggests that an acute or subacute nerve lesion is present, although a limited chronic nerve lesion cannot be excluded based on that observation alone. A clinical examination should be repeated at least every six months to assess progression, muscle weakness, upper motor neuron signs and other findings, such as bilateral wasting of the tongue, the “split hand”, head drop, emotionality and cognitive or behavioral impairment1
It is also very important to rule out any possible metabolic disorder, as toxic conditions. Earl Grey tea intoxication has been reported as a cause of widespread fasciculations and cramps2
Electromyography (EMG) is the recording of the electrical activity of the muscles. It supports the clinical suspicion and helps in the topographic diagnosis. If ALS is suspected, a systematic examination of clinically uninvolved muscles has to be done for 2 minutes as fasciculations are the hallmark of this condition. As fasciculation potentials in ALS and benign fasciculation syndrome are indistinguishable on grounds of waveform parameters3 and there is not a reliable biological marker of the disease, a minimum follow-up of 6 months is required before setting a prognosis. When non-progressive isolated fasciculations of the tibialis anterior muscle, it has to been examined the 5th lumbar root and the deep peroneal nerve, as localizer sensory symptoms may be absent4, and to rule out any more diffuse neurogenic processes.
Magnetic resonance imaging (MRI) is supportive to EMG findings as it is very sensitive in detecting anatomic changes that could be responsible for the radiculopathy, but there are other causes of radiculopathy besides nerve root compression. Moreover, lumbar disk protrusions can be found in asymptomatic patients independent of age5. Therefore, MRI is not appropriate if pain or foot drop are not present.
Finally, an isolated chronic left L5 radiculopathy was diagnosed related to lumbar spondyloarthrosis.
Hospital acquired infections (HAI) are one of the most common complications involving hospital care and are the leading cause of death in U.S. Central line associated Blood stream Infection (CLABSI), Ventilator Associated Pneumonia (VAP), Surgical site infection (SSI) and Catheter associated urinary tract infection (CAUTI) represent 75% of all HAI1 . HAI prevention is one of the 20 ‘priority areas’ identified in the Institute of Medicine (IOM) 2003 report ‘transforming health care quality’2. Certain HAI are preventable, but as the prevention efforts become more defined, there remains a lack of evidence of a strong return of investment for hospitals and health care payers in preventing these infections. This lack of evidence presents potential obstacles in advancing efforts to prevent infections.
Central Line Associated Blood Stream Infection (CLABSI)
CLABSI is a primary blood stream infection that develops in a patient with a central line in place within the 48 hour period before the onset of blood stream infection, which is not related to infection at another site. Central line associated blood stream infection occurs up to 80,000 times per year resulting in 28,000 deaths among patients in the Intensive Care unit (ICU). Average cost of CLABSI is approximately $ 45,000 per incidence3. CLABSI reduction is also one of the success story of how inexpensive interventions, grouped as a checklist could reduce the rate of nosocomial infections to a median rate of zero. Although quality control interventions in many areas of ICU have been studied, the idea of integrating quality indicators with group of interventions known as bundles has been validated in the ICU most successfully in CLABSI. The landmark study on reduction of CLABSI was the ‘Keystone ICU’ project funded by the Agency for Health care Research and Quality (AHRQ) 4. One hundred and three ICUs in Michigan participated in this state wide safety initiative. The study intervention recommended five evidence based procedures that were identified as having the greatest effect on the rate of catheter related BSI and the lowest barriers to implementation. The interventions were remarkably successful, nearly eliminating CLABSI entirely in most ICUs over an 18 month follow up period.
Although in short term intensive training and monitoring can lead to improved outcomes, in long term the biggest impact on decreasing HAI, is of the safety climate of the unit. Studies have linked safety climate to clinical and patient outcomes in addition to showing that the safety climate is responsive to interventions. A large study targeting the culture of safety was a follow up of the Michigan Keystone study. The study was a prospective cohort study to improve quality of care and safety culture by implementing and evaluating patient safety interventions in participating ICUs and showed large scale improvements in safety climate among diverse organizations5. As part of the national effort to reduce the HAI, the Department of Health and Human Services (HHS) launched the HHS action plan to reduce the health care associated infections in 2009. The project was titled ‘On the cusp: Stop BSI’, designed to apply the principles of comprehensive unit based safety program (CUSP) to improve the culture of patient safety and implement evidence based best practices to reduce the risk of infection. The initiative ultimately reduced mean rates of CLABSI in participating units by an average of 40%, preventing more than 2000 CLABSI, saving more than 500 lives and avoiding more than $34 million in excess health care costs6.
Ventilator Associated Pneumonia
Optimizing the care of mechanically ventilated patients is an important goal of health care providers and hospital administrators. An easily acquired and reliable marker for medical quality has been elusive for this patient population. VAP has historically been used as a marker of the quality of care associated with mechanically ventilated patient and is associated with worse outcomes7. However the diagnosis of VAP is non-specific, the clinical diagnosis by the widely used American College of Chest Physicians (ACCP) criteria includes a new progressive consolidation on chest radiography plus at least two of the following clinical criteria: fever > 38, leucocytosis or leucopenia and purulent secretions. Unfortunately, all these findings alone or in combination can occur in other non-infectious conditions, making the diagnosis of VAP subjective and prone to bias. In fact, for the last many years, the surveillance rates of VAP are decreasing, whereas the clinical diagnosis of VAP and tracheobronchitis as well as antibiotic prescribing remains prevalent. External reporting pressures may be encouraging stricter interpretation of the subjective signs that can cause artifactual lowering of the VAP rates. The result is that, it is almost impossible to detangle the relative contribution of quality improvement efforts in the ICU versus surveillance efforts as explanation for the currently observed lower rates of VAP8.
To eliminate the subjectivity and inaccuracy and to create an objective , streamlined and potentially automatable criteria, Center of Disease Control (CDC) now recommends surveillance of ventilator associated events (VAE) as a more general marker and defines it as sustained increase in patient’s ventilator settings after a period of stable or decreasing support . There are three definition tiers within the VAE algorithm; 1) Ventilator Associated Condition (VAC); 2) Infection Related Ventilator Associated Complication (IVAC); and 3) Possible and probable VAP. The screening for VAC captures a similar set of complications to traditional VAP surveillance, but it is faster, more objective and potentially a superior predictor of clinical outcomes9. In a CDC funded study of 597 mechanically ventilated patients on use of VAC as an outcome predictor, it was noted that 9.3% of the study population had a VAP, whereas 23% had VAC. VAC was associated with increased mortality (odds ratio of 2.0) but VAP was not. VAC assessment was also faster (mean 1.8 minutes vs 3.9 minute per patient) 10.
Similar to the CLABSI bundles, prevention of VAP by utilization of evidence-based bundles of care has proved to be a very successful. Heimes and colleagues recently conducted a study examining 696 consecutive ventilated patients in a level 1 trauma center to evaluate a VAP prevention bundle with 7 elements. They found a VAP rate of 5.2/1000 days of ventilator support in the pre intervention phase, while a 2.4 /1000 and 1.2/1000 days (p= 0.085) in the implementation and enforcement periods respectively11.
Health care associated UTI account for up to 40% of infections in hospitals and 23% of the infections in the ICU. The vast majority of UTIs are related to indwelling urinary catheters. CAUTI result in as much as $ 131million excess direct medical costs nationwide annually12. Since October 2008, Center of Medicare Services (CMS) no longer reimburses hospitals for the extra costs of managing a patient with hospital acquired CAUTI.
There are certain factors like Diabetes mellitus, old age or severe underlying illness that places patients at a greater risk of CAUTI, but there also are modifiable factors like non adherence to aseptic catheter care recommendations and duration of catheterization that can be targeted by quality improvement efforts, to decrease the risk13. The key strategies for prevention of CAUTI include avoiding insertion if possible, early removal by implementation of checklists, nurse based interventions or daily electronic reminders, utilization of proper techniques for insertion and maintenance and considering alternatives to indwelling catheters like intermittent catheterization, condom catheters and portable bladder ultrasound scanner. Most of these strategies have been utilized in quality improvement efforts to decrease CAUTI. Assessment of the need is essential as Munasinghe et al have found urinary catheter placed in 21 to 50% of patients for inappropriate reasons14. A nurse based reminder to physician to remove unnecessary urinary catheters in a Taiwanese hospital resulted in reduction of CAUTI from 11.5 to 8.3 /1000 catheter days15. Similarly utilization of electronic urinary catheter reminders system and stop orders have been shown to reduce the mean duration of catheters by 37% and CAUTI by 2%16. Utilization of condom catheter has also been shown to be effective in reducing bacteriuria, symptomatic UT and mortality as compared to indwelling catheter17.
Final word
Health care is often compared with airline industry with six sigma efficiency. This would translate to 0.002 defective parts or errors/million, obviously we are not close to that and may not be realistic. However this also cannot be an excuse to rationalize poor practice culture. As in any industry, in health care to establish change it is essential to regulate interpersonal interactions. With behaviors change leading to changes in processes of care, change is not only possible, it is sustainable.
A 26-year-old previously healthy male presented with a two day history of pain in his left wrist following trauma inflicted while playing volleyball. It was aggravated by movements around the affected joint. Clinical examination revealed mild tenderness over the left wrist with full range of movements and absence of any swelling. Distal neurovascular status was intact. X-ray of the left hand and wrist was done to rule out an injury to the bones (Fig. 1). Fig. 1: X-Rays of left hand and wrist
What diagnosis does the X-ray findings indicate?
Fracture of left scaphoid bone
Osteoblastic metastases
Osteopathia striata
Tuberous sclerosis
Osteopoikilosis
Answer / Discussion
The X-ray in Fig. 1 shows multiple small hyperdense oval and circular lesions scattered in all small bones of the left hand, with preservation of cortical thickness. These findings are suggestive of osteopoikilosis. Similar lesions were also present in the contralateral hand and wrist, as well as the pelvis (Fig. 2), on X-rays done subsequently.
Fig. 2: X-Ray Pelvis showing bone islands
Patient was counselled and reassured about the radiological findings. He was prescribed oral Paracetamol and topical Piroxicam for three days and asked to rest the affected joint. Osteopoikilosis (also called spotted bone) is a benign, possibly autosomal dominant dysplasia of bones, occurring in 1 per 50,000 people.1 Small bones of hand and feet, long tubular bones and pelvis are most frequently affected. The condition is asymptomatic and is diagnosed incidentally on radiographs taken for other problems. The diagnosis is straightforward, based on the typical radiological appearances of small (up to 10mm) hyperdense opacities distributed symmetrically. No further investigations or any specific treatment are indicated. Patients need to be reassured about the benign nature of radiological findings.
Osteoblastic metastases occur in the older age group, are generally larger in size and do not have such a uniformly symmetric distribution. Osteopathia striata is another rare bone dysplasia, characterized by long hyperdense striations mainly in the metaphyses of long bones and pelvis.2 Sclerotic bone lesions in tuberous sclerosis are frequently seen in the axial skeleton especially calvarias and spine, are at times distributed focally and have irregular borders and variable size.3 Subperiosteal new bone formation may be present and other clinical features like epilepsy may also provide a clue. As seen in Fig. 1, there is no break in the continuity of scaphoid bone, thus ruling out a fracture.
Hyperthyroidism is a common endocrine disorder and is mainly treated with anti-thyroid medications like propylthiouracil (PTU) and carbimazole. These medications have a large number of adverse effects, the commonest being skin rashes, and some are rare like agranulocytosis. Vasculitis is uncommon, but ANCA positivity is reported more in propylthiouracil and rarely with carbimazole or methimazole (1).We report a female patient with Graves’ disease who developed ANCA associated vasculitis while on carbimazole treatment.
Case report
A 29 year old female Filipino patient came to us with history of palpitations, tremors and weight loss for the last one month. Her thyroid profile showed severe hyperthyroidism (TSH <0.005, FT3-11.5, FT4-45.6) She was diagnosed with Graves’ disease as her anti-TSH receptor positive and was started on carbimazole 10mg tds. After three weeks of treatment, she developed macular rash over arms and legs and swelling of small joints of both hands. She noticed pain and colour change of both the hands and experienced typical Raynaud’s phenomenon. She had no renal or lung involvement.
On examination her blood pressure was 120/84mmHG, pulse 104 beats per min, temperature -37.1̊C. She had a mild diffuse goiter. Her X-ray chest, ECG and urine dipstick routine were all normal. Her CRP and ESR were raised. X-rays of the hands were normal. P-ANCA was positive. Antimyeloperoxidase antibody was positive. Anti-TPO and TSH receptor antibodies were positive.
Diagnosis of carbimazole induced vasculitis was made. The patient was treated with prednisolone 40mg daily once daily which was tapered over three weeks. She improved within 48hours and was asymptomatic after three weeks. She was treated successfully with radioiodine ablation. Her MPO-ANCA after 6 months was negative.
Figure 1. Pictures of the hands showing Raynaud’s phenomenon
Figure 2. Pictures of the hands showing Raynaud’s phenomenon
Discussion
ANCA positive vasculitis in association with antithyroid drugs was first reported in 1992 (2).There has been 32 cases of ANCA positive vasculitis associated with antithyroid medications reported up until now (3). The presenting symptoms are variable and may include renal involvement (67%), arthralgias (48%), fever (37%), skin involvement (30%), respiratory tract involvement (27%), myalgias (22%), scleritis (15%) and other manifestations (18%) (3).
In these patients the underlying thyroid disease is most commonly Graves’ disease but ANCA positive vasculitis has also been seen with association with toxic multinodular goitre (4). Recent studies have shown high frequency of ANCA positivity in patients with Graves’ disease treated with antithyroid medications, especially with PTU. Most cases of ANCA positivity are seen in patients on long term therapy (greater than 18 months) or in those with recent commencement of therapy as seen in our patient. However, a small percentage of these go on to develop features of vasculitis (3).
The majority of cases of vasculitis (88%) have been reported in association with PTU, vasculitis associated with carbimazole is very rare (5, 6, and 7). The pathogenesis of ATD associated vasculitis is not clearly understood. PTU has been shown to accumulate within neutrophils (8) and bind myeloperoxidase (9). The binding alters the configuration of myeloperoxidase (9) and may promote formation of autoantibodies in susceptible people. There has been no data with regards to whether carbimazole can alter the configuration of myeloperoxidase. ANCA positive vasculitis may be more common in patients of Asian ethnic origin, with half of cases reported from Japan (3). Our patient was from Philippines.
Wadw et al have reported 25% of patients were positive for MPO-ANCA in PTU group whereas in methimazole group 3.4% were positive (10).
This case highlights the awareness of this relatively rare adverse effect of a thyroid medication which may lead to fatal renal and pulmonary complications. Early diagnosis and withdrawal of the offending medication is important. In asymptomatic patients the significance of ANCA positivity is not clear but early definitive therapy in the form of radioiodine ablation or surgery should be considered.
A 44-yr-old patient with history of ileal Crohn’s disease was admitted to our Department because of asthenia, subclinical jaundice, painful hepatomegaly, fluid retention and ascites. In 2008 the patient was diagnosed with bladder cancer and was treated by surgical resection of the cancer and intravesical chemotherapy with mitomicyn C. In 2010 he was given azathioprine (AZA) at 2 mg/kg for Crohn’s disease and 3 months later he developed an increase in serum alkaline phosphatase, gamma-glutamyl transpeptidase and transaminases. He was then started on 1.5 mg/kg 6-mercaptopurine (6-MP) once daily. After 9 months he stopped 6-MP because of nausea, vomiting and abnormal liver function tests; 6-MP was therefore discontinued until the normalisation of markers of liver function. Two months later, when the transaminases were within the normal range, he received 6-thioguanine (6-TG) 25 mg a day, that was progressively increased to 80 mg a day. Three months later, the patient was referred to our Department with painful hepatomegaly, ascites and asthenia. Laboratory tests on admission revealed an elevation in AST 198 U/l and ALT 209 U/l. Total bilirubin was 3 mg/dl (direct bilirubin 1.5 mg/dl), LDH 784 U/l, alkaline phosphatase 191 U/l and ammonia 112 umol/l. Virological markers (HBsAg, HBcAb, anti HCV, HBV DNA) were negative. Patient was apyrexial, showed normal blood pressure (130/80 mmHg), tachycardia (110 bpm) and 97% SaO2 on room air. Physical examination revealed right hypochondrial tenderness, abdominal distension and shifting dullness, suggesting the presence of ascites. The rest of the physical examination was unremarkable. An echo-Doppler evaluation revealed thin linear suprahepatic veins and confirmed the presence of ascites. A CT scan of the abdomen showed hepatomegaly with dishomogeneous enhancement after dye injection (mosaic pattern). There was no evidence of any venous thrombosis or splenomegaly (Figure 1A); 6-TG was withdrawn empirically and the patient was started on therapy with albumin 25 g/day and spironolactone 200 mg/day. The average serum Na+ level during diuretic treatment was 134 mEq/l. An abdominal paracentesis of two litres was necessary, due to the progressive increase of ascites.
FIGURE 1A. CT scan of the abdomen on admission: Dishomogeneous enhancement of the liver after dye injection (mosaic pattern) (arrow). Suprahepatic veins are not detectable.
FIGURE 1B. Histological pattern of the liver biopsy specimen: marked centrilobular congestion (arrows) with hepatocyte dropout. There is no evidence of centrolobular veins thrombosis.
A routine laboratory investigation of ascitic fluid showed < 500 leukocytes/µL and < 250 polymorphonuclear leukocytes (PMNs)/µL. The ascitic fluid total protein level was 2.1 g/dl and serum-ascites albumin gradient (SAAG) was > 1.1 g/dL. No neoplastic cells were found. A transjugular liver biopsy was then performed, showing marked centrilobular hemorrhage with hepatocyte necrosis. There was mild ductular reaction, with no evidence of centrilobular vein thrombosis. The histologic diagnosis confirmed veno-occlusive disease (VOD) (Figure 1B). Screening for thrombophilia was also done, showing low levels of serum protein C and protein S. There was no mutation of JAK-2 V617F. The patient was then treated with a hyposodic diet, mild hydric restriction, enoxaparin,spironolactone, lactulose and omeprazole. He was discharged two weeks later, and after 3 months a complete regression of ascites and hepatomegaly occurred, and echography of the liver was unremarkable (Figure 2A and 2B).
FIGURE 2A. Echography of the liver at follow up. No evidence of ascites.
FIGURE 2B. Echography of the liver at follow up. No evidence of ascites. Suprahepatic veins are detectable (arrow)
Discussion
Although VOD was known among complications of 6-TG in childhood, this case-report emphasises the occurrence of VOD in adults with Crohn’s disease, as first described by Kane et al. in 20041. The thiopurine drugs were developed more than 50 years ago, and 6-MP was first used as a drug in 19522. Since then, 6-MP and 6-TG have been widely used to treat acute lymphoblastic leukemia in children. VOD mimicking Budd-Chiari like disease was then described as a frequent complication of 6-TG in pediatric patients given the drug for lymphoblastic leukaemia. Later on, in 1976, Griner et al. described the cases of two adult male patients with acute leukaemia developing a fatal Budd-Chiari-like disease while receiving 6-TG3. Since patients were given 6-TG plus cytosine arabinoside, authors were unable to ascribe this complication solely to 6-TG3. VOD exclusively related to 6-TG was first described by Gill et al., who observed a clinically reversible liver VOD developing in a young man with acute lymphocytic leukemia after 10 month administration of 6-TG4. Furthermore, sinusoidal obstruction was also reported in a patient with psoriasis treated with 6-TG and other cytotoxic therapy5. In 2006, a European 6-TG Working Party established that 6-TG should be considered a rescue drug in stringently defined indications in inflammatory bowel diseases (IBD). The indication for administration of 6-TG should only include its use for maintenance therapy as well as intolerance and/or resistance to aminosalicylates, azathioprine, 6-mercaptopurine, methotrexate and infliximab. Moreover, 6-TG must be withdrawn in case of overt or histologically proven hepatotoxicity6. Although Ansari et al 7 found no nodular regenerative hyperplasia (NRH) in the liver of patients given 6-TG, Dubinsky et al.8 described NHR as a common finding in 6-TG-treated patients with inflammatory bowel disease in the absence of VOD. By contrast, in our case report we showed histological pattern of VOD and, in accord with Gisbert et al.9, would suggest that 6-TG should not be administered out of a clinical trial setting. Given that the proportion of patients with Crohn’s disease achieving an improvement of symptoms during 6-TG treatment is similar to that after methotrexate10 or infliximab6, these drugs should therefore be considered as second line therapy in patients intolerant or resistant to azathioprine and 6-mercaptopurine.
The number of individuals surviving cancer is expected to rise by one-third according to estimates from the American Cancer Society and the National Cancer Institute1. This means that in the UK over 3 million individuals, and in the USA over 18 million individuals, will be living with the consequences of cancer by 2,022. The increase in the number of survivors is attributed to earlier diagnosis, an aging population, better cure rates and more effective systemic therapies to keep patients with metastatic disease alive for longer. To achieve these benefits, patients often have to endure more complex and arduous therapies, frequently leaving them beleaguered with acute and long-term adverse effects. In addition to being unpleasant, these adverse effects result in financial implications for patients and their families, as well as resulting in a greater usage of health resources.
Although the importance of exercise is beginning to be recognised by health professionals, advocacy groups and charities, it still remains an under-utilised resource. This article highlights the evidence that a physically active lifestyle and formal exercise programmes can help relieve many of the common concerns and adverse effects which plague individuals in the cancer survivorship period.
Physical activity improves well-being after cancer
Dozens of interventional studies have tested the feasibility and potential benefits of exercise in cancer survivors2,3,4. Recent meta-analyses of randomised trials involving exercise interventions after cancer, encouragingly demonstrate that the benefits of exercise spanned across several common cancer types and following a range of treatments including surgery, radiotherapy, chemotherapy, hormones and even the newer biological therapies. The most recent meta-analysis of 34 randomised trials published in the BMJ in 2012 involving patients exercising after cancer, demonstrated a benefit for a number of troublesome symptoms particularly fatigue, mood, anxiety and depression; muscle power, hand grip, exercise capacity and quality of life5.
The American College of Sports Medicine also published a comprehensive review of exercise intervention studies in cancer populations which included data from 85 RCT’s of exercise in cancer survivors. Evidence consistently demonstrated that exercise could be performed safely in adjuvant and post-treatment settings. Exercise led to significant improvements in aerobic fitness; increased flexibility and strength; quality of life; anxiety and depression; fatigue, body image, size and composition4.
The individual categories of symptoms which commonly afflict cancer survivors are now discussed in more detail:
Cancer related fatigue (CRF) is one of the most distressing symptoms experienced by patients during and after their anti-cancer therapies. It is reported by 60-96% of patients during chemotherapy, radiotherapy or after surgery, and by up to 40% of patients taking long-term therapies such as hormonal or biological therapies6. The first step to treating CRF is to correct, if possible, any medical conditions that may aggravate it, such as anaemia, electrolyte imbalance, liver failure and nocturia; or to eliminate drugs such as opiates, anti-histamines and anti-sickness medication7. The role of exercise was reviewed in 28 randomised, controlled trials (RCTs) involving 2083 participants in a variety of exercise programmes and showed that exercise improved CRF, although the benefit overall was small8. A second review of 18 RCTs involving 1,109 participants, sub-divided the data into types of exercise and demonstrated that supervised exercise programmes had the most impact on CRF9. Further meta-analyses and reviews have also showed that supervised exercise programmes had better results, with a greater reduction in CRF amongst breast cancer survivors assigned to exercise programmes compared to home-based programmes4,5,8,10.
Psychological distress, including anxiety and depression, is common after cancer with reported prevalence rates of 25-30%11. Patients with psychological distress have also been shown to have reduced survival compared to those who are psychologically healthy12. Exercise may help alleviate this symptom and improve mood, as a number of observational studies have shown that cancer patients who exercise have reduced levels of depression and anxiety, better self-esteem and are happier, especially if they involve group activities13. The recent meta-analyses of RCTs also demonstrated a reduction in anxiety and depression among individuals assigned to exercise programmes4,5.
Quality of life (QOL) is lower in many cancer sufferers and survivors, linked to other physical and psychological symptoms of cancer and its’ treatment. Meta-analyses of studies of exercise intervention programmes have demonstrated an improvement of QOL at all stages of the illness for the common cancer types and following several types of treatment4,5. For example, in a study involving 1,966 patients with colorectal cancer, patients achieving at least 150 minutes of physical activity per week had an 18% higher QOL score than those who reported no physical activity, as measured by the QOL FACT-C14. Another study showed similar benefits for breast cancer survivors who had completed surgery, radiotherapy or chemotherapy, and also demonstrated that change in peak oxygen consumption correlated with change in overall QOL15.
Weight gain:45% of women with breast cancer report significant weight gain16, and in a study of 440 prostate cancer survivors, 53% were overweight or obese17. For patients with bowel cancer, the CALBG 8980 trial showed that 35% of patients post-chemotherapy were overweight (BMI 25.0–29.9), and 34% were obese (BMI 30.0–34.9) or very obese (BMI >35)16. The reasons for this are multifactorial, but may include other symptoms of cancer treatment such as fatigue and nausea, causing patients to stop exercising. Regardless of the reasons for weight gain, numerous reviews and a comprehensive meta-analysis of the published literature have demonstrated that individuals who gain weight after cancer treatments have worse survival and more complications18. Fortunately, supervised exercise programmes have been shown to reduce weight and have significant other benefits on body constitution and fitness, such as improved lean mass indices, bone mineral density, cardiopulmonary function, muscle strength and walking distance18,19.
Bone mineral density (BMD): Pre-menopausal women who have had breast cancer treatment are at increased risk of osteoporosis, caused by reduced levels of oestrogen brought on by a premature menopause due to chemotherapy, surgery or hormones. Men who receive hormone deprivation therapy for prostate cancer are also at an increased risk of developing osteoporosis. Accelerated bone loss has also been reported for many other cancers, including testicular, thyroid, gastric and CNS cancers, as well as non-Hodgkin’s lymphoma and various haematological malignant diseases20,21. Lifestyle factors linked to an increase in the risk for developing osteoporosis include a low calcium and vitamin D intake, a diet low in plant-based protein, lack of physical activity, smoking and excessive alcohol intake22. A number of studies have linked regular physical activity with a reduction in the risk of bone mineral loss. Sixty six women with breast cancer were randomized to a control group or an exercise programme. The rate of decline of BMD was -6.23% in the control group, -4.92% in the resistance exercise group, and -0.76% in the aerobic exercise group. The statistically significant benefit was even greater in pre-menopausal women23. In another RCT of 223 women with breast cancer, it was found that exercise, over 30 minutes 4-7 times a week, helped preserve bone mineral density even when bisphosphonates (risedronate), calcium and vitamin D had already been prescribed24.
Thromboembolism: Those with pelvic involvement, recent surgery and immobility, previous history of varicose veins or thrombosis or receiving chemotherapy, are at higher risk25. Although strategies such as compression stockings, warfarin and low molecular weight heparin are essential, early mobilisation and exercise remains a practical additional aid in reducing this life-threatening complication18,26.
Constipation caused by immobility, opiate analgesics or anti-emetics during chemotherapy is a significant patient concern. Exercise reduces bowel transit time, and ameliorates constipation and its’ associated abdominal cramps26.
Physical activity improves survival and reduces relapse
In addition to improving the side effects of treatment for cancer, regular physical activity during and after cancer appears to improve overall survival and reduces the probability of relapse. One of the most convincing studies was an RCT in which 2,437 post-menopausal women with early breast cancer were randomised to nutritional and exercise counselling, or no counselling, as part of routine follow-ups19. In the group receiving counselling, fewer women relapsed and overall survival was greater in the oestrogen-negative subgroup. In another RCT, men with early prostate cancer were randomised to an exercise and lifestyle intervention or standard active surveillance. The average PSA in the intervention group went down, whilst in the control group it went up27. This supports a previous RCT of which the primary end point evaluated a salicylate-based food supplement, but it required men in both arms to receive exercise and lifestyle counselling. Although there was no difference in the primary end point, 34% of men, who’s prostate specific antigen (PSA) was climbing before trial entry, stabilized28.
The majority of the other published evidence for a reduced relapse rate and improved survival after cancer originates from retrospective analysis or prospective cohort studies. The National Cancer Institute, in a recent meta-analysis, reviewed 45 of these observational studies. The strongest evidence was demonstrated for breast cancer survivors; the next strongest evidence was for colorectal cancer survivors, followed by prostate cancer10. The most notable are summarised below:
Breast cancer: The five most prominent prospective cohort studies (in aggregate more than 15,000 women), have examined the relationship between physical activity cancer and prognosis:
Irwin et al. (2008)29 investigated a cohort of 933 breast cancer survivors and found that those who consistently exercised for >2.5 hours per week had a 67% lower risk of all deaths compared to sedentary women.
Holmes et al. (2005)30 performed a separate evaluation of 2,987 women in the Nurses’ Health Study and found that women walking >3 hours a week had lower recurrence rates, and better overall survival.
Holick et al. (2008)31 performed a prospective observational study of 4,482 breast cancer survivors, and found that women who were physically active for >2.8 hours per week had a 35-49% lower risk of dying from breast cancer.
Pierce et al. (2007)32 found that the benefits of 3 hours of exercise were even greater if combined with a healthy diet.
Sternfeld et al. (2009)33 in the LACE study, evaluated 1,870 women within 39 months of diagnosis. There was a significant difference in overall death rate between the highest and lowest quartile of exercise levels.
Colorectal cancer: The scientific community eagerly awaits the results of the CHALLENGE RCT mentioned above, but a number of retrospective analyses of randomised chemotherapy and cohort trials have been published:
Haydon et al. (2006)34retrospectively analysed a RCT involving patients with stage III bowel cancer and found a significant association between exercise and a 31% reduction in relapse rate.
Giles et al. (2002)35found that of 526 patients recruited into the Australian Cohort Study, those participating in recreational sport 1-2 days per week had a 5 year overall survival of 71%, as opposed to 57% in non exercisers.
Meyerhardt et al. (2006)16 found in an analysis of the Intergroup CALGB study, that physically active patients with bowel cancer had 35% reduction in relapse rate in after chemotherapy.
Meyerhardt et al. (2009)36 analysed 668 patients with colorectal cancer within the Health Professionals Study. Men who exercised >27 vs. < 3METS-hours / week had a lower cancer-specific mortality.
Prostate cancer: Three cohort studies have demonstrated a survival benefit for physically active men with prostate cancer:
Kenfield et al. (2011)37performed a subset analysis of 2,686 men with prostate cancer, within the Health Professional Study, who exercised >30minutes per week or >3 MET-hours of total activity, had a 35% lower risk of overall death, and men who walked at a brisk pace for >90 minutes had a 51% lower risk of overall death.
Richman et al. (2011)38 reported that 1,455 men with prostate cancer, walking more than 3 hours a week, correlated with an improved survival but only if >3miles/hour.
Giavannucci (2005)39, within a prospective analysis, reported that men who exercised vigorously had a lower risk for fatal prostate cancer, although this effect was only seen for men over the age of 65.
Quantity and type of exercise recommended for cancer patients
For reduced cancer relapse and improved well-being, most of the cohort studies summarized above suggest moderate exercise of around 2.5 to 3 hours a week for breast cancer survivors. However, for prostate cancer survivors, mortality continues to decrease if the patient walks 4 or more hours per week, and more vigorous activity is also associated with significant further reductions in risk for all-cause mortality37. When the mode of exercise is primarily walking, a pace of at least 3 miles/hour (for >3 hours/week) is recommended for a reduced risk of relapse 38. Therefore, both the pace and duration of exercise affect the survival benefit achievable from exercise, with more vigorous activity generally having a greater benefit (see Table 1). The best results appear to be with programmes including a combination of aerobic and resistance exercises, particularly within a social group.
Table 1: Summary of exercise guidelines for cancer survivors
· Exercising for >3 hours/week has proven benefits for cancer survival
· A pace of at least 3 miles/hour when walking provides greater benefit than a slower pace
· For optimal benefit, exercise should consist of a combination of resistance and aerobic exercises
· Supervised exercise programmes have shown greater benefits for cancer survivors than home-based programmes
The precise amount of exercise has to be determined on an individual basis and depends on pre-treatment ability, current disability caused by the cancer itself or the treatment, as well as time proximity to major treatments. An exercise programme supervised by a trained professional has major advantages, as they can design a regimen which starts slowly and gradually builds up to an acceptable and enjoyable pace. In addition, they can help motivate the individual to continue exercising for the short and the long-term, and they can judge the optimal exercise levels to improve fatigue, and not aggravate it.
The underlying mechanisms of the potential anti-cancer effects of exercise
The body’s chemical environment significantly changes after exercise, best demonstrated in the Ornish study, which found that serum from prostate cancer patients who exercised, had an almost eight times greater inhibitory effect on the growth of cultured androgen dependent prostate cancer cells compared to serum from patients in the control group27. The precise chemical mechanism, which the anti-cancer effect remains incompletely understood, but one of the most likely mechanisms involving growth factors such as insulin-like growth factor (IGF-1) and its’ binding proteins insulin-like growth factor binding proteins (IGFBPs), due to the central role of these proteins in the regulation of cell growth (see Table 2). After binding to its receptor tyrosine kinase, IGF-1 activates several signalling pathways including the AKT pathway, leading to the inhibition of apoptosis and the promotion of cell growth and angiogenesis34,40,41. An inverse relationship of cancer risk with IGFBP3 levels has also been shown, although this effect has not been confirmed in all studies42. Exercise has been shown to increase the levels of IGFBP3, and this was associated with a 48% reduction of cancer-specific deaths in a large prospective cohort study of 41,528 participants43. Decreased levels for IGF-1 in physically active patients have been reported with an associated survival benefit44.
Table 2: Summary of the potential biochemical pathways of the anticancer effects of exercise
Class of Effector Molecule
Effector Molecule
Effects of Exercise on Effector Molecule
Cell growth regulators
IGF1
Decreased levels
IGFBP3
Increased levels
Proteins involved in DNA damage repair
BRCA1
Increased expression
BRCA2
Increased expression
Regulator of apoptosis and cell cycle arrest
p53
Enhanced activity
Hormones
Oestrogen
Decreased levels
Vasoactive intestinal protein (VIP)
Decreased levels
Leptin
Decreased levels (indirect)
Immune system components
NK cells
Enhanced activity
Monocyte function
Enhanced activity
Circulating granulocytes
Increased proportion
Exercise has also been shown to have a large impact on gene expression, although the mechanisms through which the patterns of gene expression are affected remain to be determined. In a recent study of the mechanisms through which exercise impacts prostate cancer survival, it was found that 184 genes are differentially expressed between prostate cancer patients who engage in vigorous activity, and those who do not 37. Amongst the genes that were more highly expressed in men who exercise were BRCA1 and BRCA2, both of which are involved in DNA repair processes.
Another neuropeptide which changes after exercise is Vasoactive Intestinal Protein (VIP). Breast and prostate cancer patients have been found to have higher VIP titres compared to individuals who regularly exercise, and who have increased production of natural anti-VIP antibodies45. In hormone-related cancers such as cancers of the breast, ovaries, prostate and testes, the association between high levels of circulating sex hormones and cancer risk is well established46. Another mechanism through which exercise may affect cancer, is through decreasing the serum levels of these hormones. For breast cancer survivors, the link between exercise and lower levels of oestrogen has been shown13,34,47. An indirect, related mechanism is that exercise helps reduce adiposity, and adiposity in turn influences the production and availability of sex hormones48. In addition, greater adiposity leads to higher levels of Leptin, a neuropeptide cytokine with has cancer promoting properties49,50.
Other pathways include the modulation of immunity, such as improvements in NK cell cytolytic activity 11; the modulation of apoptotic pathways through impacting on a key regulator, p5351, and an exciting recent discovery, the messenger protein irisin, which is produced in muscle cells in response to exercise and is found is to be an important molecule in linking exercise to the health benefits52 , However, we are only beginning to scratch the surface with these and the other mechanisms discussed here, and much more research needs to be done to in this area.
Incorporating exercise into mainstream cancer management
The challenge for health professionals is how to encourage and motivate individuals with cancer to increase their exercise levels. Some, of course, are motivated to increase physical activity or remain active after cancer. However, a recent survey of 440 men with prostate cancer found that only 4% of patients exercised for more than the 3 hours a week recommended by the WCRF17. Macmillan Cancer Relief has produced a series of helpful booklets and web-based patient information materials designed to inform and motivate individuals to exercise as part of its ‘Move More’programme. The Cancernet website has a facility to search for local exercise facilities by postcode, which can be an aid for health professionals when counselling patients. It highlights activities that men will hopefully find feasible and enjoyable such as golf, exercise groups and walking groups, and are encouraged to attend in addition to work place activity and gardening.
Several pilot schemes have been started throughout the UK with the aim to incorporate exercise programmes into standard oncology practice. The difficulty with small schemes is that they tend to be poorly funded, often poorly attended and are unlikely to be sustainable in the longer term. Many agree that the gold standard model would be similar to the cardiac rehabilitation programme53. This would involve a hospital scheme run by a physiotherapist or an occupational therapist, supervising patients immediately after surgery, radiotherapy and even during chemotherapy. This is followed by refering the patient to a community-based scheme for the longer term. Unfortunately, this type of scheme is expensive and unlikely to be funded at present, despite the obvious savings by preventing patient relapsing and ultilising health care facilities to help late effects of cancer treatment54. However, expanding existing services, such as the National Exercise Referral Scheme, is a practical solution. The National Exercise Referral Scheme exists for other chronic conditions such as cardiac rehabilitation, obesity and lower back pain. The national standards for the scheme to be expanded to include cancer rehabilitation were written and accepted in 2010. Training providers have now developed training courses for exercise professionals set against these standards. Trainers completing the course gain a Register of Exercise Professionals (REPs) Level Four qualification, allowing them to receive referrals from GPs and other health professionals.
Conclusion
There are a wealth of well-conducted studies which have demonstrated an association between regular exercise and lower risk of side effects after cancer, as well as reasonable prospective data for a lower relapse rates and better overall survival. However, as there are several overlapping lifestyle factors, which are difficult to investigate on their own, there remain some concerns that exercisers may do better in these studies because they are less likely to be over-weight, more likely to have better diets and to be non-smokers. Although the existing RCTs provide encouraging evidence that exercise intervention programmes are beneficial, further large RCTs are needed, particularly in terms of cost-effectiveness, before commissioner’s start investing more in this area.
The American Diabetic Association (ADA) and the American College of Endocrinology (ACE) recommend HbA1c levels as diagnostic criteria for diabetes mellitus. Physicians have adopted HbA1c levels as a convenient way to screen for diabetes, as well as to monitor therapy. There exists concern that because HbA1c is formed from the glycation of the terminal Valine unit of the β-chain of haemoglobin, it may not be an accurate surrogate to ascertain glycemic control in certain conditions that affect the concentration, structure and function of haemoglobin. It makes logical sense to infer that HbA1c levels should at least in part reflect the average haemoglobin concentration ([Hb]). Kim et al (2010) stated that iron deficiency is associated with shifts in HbA1c distribution from <5.0 to ≥5.5% 1 and significant increases was observed in the patients' absolute HbA1c levels 2 months after treatment of anaemia.2 There is a dearth of literature on HbA1c levels in the anaemia population, and a reference range for this unique population does not currently exist. There are a few documented studies on this matter, the findings of which are at best, inconsistent.
It is thought that the various types of haemoglobin found in the myriad of haemoglobinopathies may affect haemoglobin-glucose bonding and/or the lifespan of haemoglobin, and by extrapolation, HbA1c level. Hence, extending target HbA1c values to certain haemoglobinopathaties may be erroneous due to potential differences in glycation rates, analytical methods (HbF interfers with the immunoassay method) and some physiological challenges (markedly decreased red cell survival).3
There is a significant positive correlation between haemoglobin concentration and HbA1c in the patients with haemolytic anaemia.4,5 Cohen et al (2008) reported that observed variation in red blood cell survival was large enough to cause clinically important differences in HbA1c for a given mean blood glucose,6 and haemolytic disorders may cause falsely reassuring HbA1c values.7 Jandric et al (2012) inferred that in diabetic population with haemolytic anaemia, HbA1c is a very poor marker of both overall glycemia and haemolysis.8 Mongia et al (2008) report that immunoassay methods for measuring HbA1c may exhibit clinically significant differences owing to the presence of HbC and HbS traits.9 However, Bleyer et al report that sickle cell trait does not affect the relationship between HbA1c and serum glucose concentration and it does not appear to account for ethnic difference in this relationship in African Americans and Caucasians.10
Koga & Kasayama (2010) advise that caution should be entertained when diagnosing pre-diabetes and diabetes in people with low or high haemoglobin concentration when the HbA1c level is near 5.7% or 6.5% respectively, citing the implication of changes in erythrocyte turnover. They further assert that the trend for HbA1c to increase with iron deficiency does not appear to necessitate screening for iron deficiency to ascertain the reliability of HbA1c in this population.11
In the light of the uncertainty in the influence of anaemia and haemoglobinopathies on HbA1c, it is imperative that clinicians are aware of the caveats with HbA1c values when they make management decisions in the anaemic population.12 There is currently a call for the use of other surrogates for ascertaining average glycemic control in pregnancy, elderly, non-Hispanic blacks, alcoholism, in diseases associated with postprandial hyperglycemia, genetic states associated with hyperglycation, iron deficiency anaemia, haemolytic anaemias, variant haemoglobin states, chronic liver disease, and end-stage renal disease (ESRD).13,14
Study objectives and hypothesis
The study attempts to discern clinical differences in HbA1c levels in patients with anaemia compared to non-anaemic population, as well as to quantify and show the direction of such difference if they indeed exist. We hypothesize that as glucose is covalently bound to haemoglobin in glycosylated haemoglobin, HbA1c levels in non-diabetic anaemic population is significantly lower than in non-diabetic, non-anaemic population.2 However, this relationship may not hold true for certain anaemias, haemoglobinopathies and hyperglycation states in some genetic syndromes.
Study design and method
The study is a retrospective chart review of patients with and without anaemia who underwent haemoglobin concentration and HbA1c level testing at The Brooklyn Hospital Center (TBHC) from July, 2009 to June, 2013. Using Cohen (1987) power table, assuming a power of 0.8, alpha level of 0.05, and a small effect size of 0.2 standard deviations (SD), sample size estimation of 461 was computed. A convenient sampling method was used to select patients who meet inclusion criteria, absent exclusionary conditions. In using this sampling method, we queried the electronic medical record at the TBHC using the below-listed inclusion and exclusion criteria. The query generated a list of “potential subjects”. We then reviewed the electronic chart of each patient on this list to confirm that they indeed meet all study criteria (excluding further if any exclusion criteria was identified on “second look”. We continued the selection until the computed minimum sample size of 461 was significantly exceeded. During this process, we had to examine every patient on the “potential subject” list generated by the initial query to achieve this goal. For the purpose of the study, anaemia is defined as haemoglobin concentration <11g/dl.
Inclusion criteria:
iStudy participant must be at least 21 years of age. We adopted this age criteria because at TBHC, electronic medical records was only available for the non-pediatric population over the study period. Patients below 21 years were managed at the pediatrics department using paper charts until the recent adoption of the EMR system. It would have been difficult conducting the study using paper charts.
iStudy participant must have at least one documented HbA1c level obtained within a month of a haemoglobin concentration assay. This criterion was adopted to allow for more inclusiveness in the study. It is our experience that haemoglobin assays may not be available on the same day as HbA1C assays considering the retrospective nature of the study.
Exclusion criteria:
Confirmed cases of diabetes mellitus (using two or more of the following: presence of symptoms related to diabetes, fasting blood glucose, 2 hours post-prandial glucose, and oral glucose tolerance test).
Documented history of gestational diabetes (GDM)
Documented history of endocrinopathy with affect for glycemic control
Current or prior use of medication with potential to increase or decrease HbA1c (includes, but not limited to antidiabetics, corticosteroids, statins, and antipsychotics)
Pregnancy or pregnancy-related condition within three months of HbA1c assay
Haemoglobin concentration <6 g/dl or >16g/dl.
Blood loss or blood transfusion within two months of HbA1c assay
The study assumed a consistent HbA1c assay method at the study center over the study period. 482 (229 anaemic and 253 non-anaemic) were selected. The study reviewed electronic medical records of selected patients, extracting data on HbA1c, fasting blood glucose (FBG), 2-hour post-prandial serum glucose (2HPPG), 2-hour oral glucose tolerance test (OGTT), haemoglobin concentration and electrophoresis, and anaemia work-up results when available. Subsequent measures of HbA1c two months after correction of anaemia was also documented and compared to pre-treatment levels.
Results and Analysis
The mean age of the anaemic and non-anaemic was 51.8 and 64.6 years respectively. Using the student’s t-test and x2 analysis respectively, the difference in mean age of both groups (anaemia and non-anaemic) was significant at p0.05 while gender distribution was similar (p>0.05), see table 1. The mean HbA1c for anaemic and non-anaemic groups was 5.35% and 5.74% respectively, amounting to a 0.4 unit difference in (8%) in mean HbA1c. This difference was statistically significant (p0.02). A significantly higher variance was observed in the anaemia group (0.79 vs. 0.64).
Table 1: Gender and age distribution and statistics
Age in years Anaemia
#(%)
Gender (M/F)
Mean Age (in yrs)
21-44
20(8.7)
17/41
45-64
76(33.2)
43/86
≥65
133(58.1)
10/32
Total
229(100.0)
70/159
64.6
Non-anaemic
21-44
64(25.3)
23/42
45-64
134(53.0)
58/81
≥65
55(21.7)
18/31
Total
253(100)
99//154
51.8
p-Values: Age=0.023, Gender=0.061
Assuming that 95% of the population is normal, computation of HA1c reference range (mean ±1.96SD) for the anaemia and non-anaemic group yielded 3.8-6.9 and 4.5-7.0 respectively. There was a significantly positive spearman correlation between [Hb] and HbA1C (r=0.28, p0.00). The mean HbA1c level and proposed reference ranges for the five anaemia subgroups (anaemia of chronic disease [ACD], iron deficiency anaemia [IDA], mixed anaemia, macrocytic anaemia and sickle-cell disease) are shown in table 2. Using one-way ANOVA analysis, the difference in the mean [Hb] and HbA1c across anaemia subtypes was not statistically significant (p0.08 and p0.36 respectively), see table 2.
Table 2: Anaemia subtypes with HbA1c statistics
Anaemia Type
#
Mean[Hb]
MeanHbA1c
95% CI (HbA1c)
Ref. range (HbA1c)
ACD
92
9.23
5.41
5.24-5.59
3.5-7.1
IDA
78
9.41
5.38
5.22-5.54
3.9-6.8
Mixed
11
9.11
5.21
4.82-5.59
3.9-6.5
Macrocytic
43
8.83
5.14
4.92-5.37
3.7-6.6
SCD
5
9.12
5.55
4.84-6.26
3.8-7.3
Anaemia (all types)
229
9.21
5.35
5.24-5.44
3.8-6.9
Non-anaemic
253
12.87
5.735
5.66-5.81
4.5-7.0
p-values: [Hb] for anaemia subtypes=0.08, HbA1C for anaemia subtypes=0.36, HbA1C anaemia vs. non-anaemia=0.02. ACD: anaemia of chronic disease, IDA: iron deficiency anaemia, SCD: sickle cell disease.
The study also examined the anaemia group to document the effect of anaemia correction on HbA1c levels. Only 62 of the 229 anaemic participants had documented [Hb] and HbA1c after interventions to correct anaemia, see table 3 and 4.
Table 3: Trend in [Hb] and HbA1c
N
Mean
SD
SEM
Change
p-Value
[Hb]1
62
9.2
1.07
0.14
[Hb]2
62
10.1
1.98
0.25
[Hb]=0.9
0.00
HbA1c1
62
5.37
0.69
0.88
HbA1c2
62
5.35
0.66
0.83
HbA1c=0.02
0.78
[Hb]1 and [Hb]2: haemoglobin concentration pre- and post- treatment for anaemia. HbA1c1 and HbA1c2: HbA1c pre- and post-treatment for anaemia
Table 4: Trend in [Hb] and HbA1c for anaemia subtypes
N
Mean[Hb]1
Mean[Hb]2
Δ Hb
pValue
MeanHbA1c1
MeanHbA1c2
ΔA1c
pVal
ACD
33
9.1
9.7
0.6
0.0
5.44
5.35
0.09
0.3
IDA
21
9.4
10.7
1.3
0.0
5.30
5.33
0.03
0.8
Mixed
1
Macrocytic
6
SCD
1
Total
62
9.2
10.1
0.9
0.0
5.37
5.35
0.02
0.8
ΔHb: change in haemoglobin concentration ([Hb]), ΔA1c: change in HbA1c
Using the student’s t-test, analysis, a 0.9g/dl mean improvement in [Hb] in the anaemia group (significant at p0.00) did not result in a statistically significant change in HbA1c (-0.02 units, p0.78). Similar results were obtained with anaemia of chronic disease and iron deficiency anaemia (ICD: change [Hb] =+0.6g/dl, change HbA1c=0.09, p0.31; IDA: change [Hb]=+1.3g/dl, change HbA1c=0.03, p0.79).
Discussion
There was an over-representation of the elderly in the anaemia group (58.1% vs. 21.7%). This is not unexpected as nutritional anaemia and anaemia of chronic disease increase in prevalence with the increasing co-morbidities associated with increasing age. The linear relationship between [Hb] and HbA1c holds true for anaemic and non-anaemia populations. There is a statistically significant difference of 0.4units (8%) in the mean HbA1c between the anaemic and the non-anaemic population. This difference is even more marked when the lower limit of the range is compared (3.8 vs. 4.5, difference of 0.7unit, 18%), the significance of which is not as clinically impacting as the upper limit of the range (diabetes mellitus diagnostic criteria). However, the relatively lower limit of normal for HbA1c in anaemic subgroups (especially of anaemia of chronic disease) may make low values of HbA1c in these patients less indicative of over-enthusiastic glycemic control, as well as less predictive of the increase in mortality associated with such tight control.
The upper range of normal for HbA1c for the anaemia and the non-anaemic groups and by extrapolation the proposed diagnostic criteria for diabetes, is however more similar (6.9 vs. 7.0%). This result appear consistent with Koga and Kasavama (2010) assertion that the trend in HbA1c does not appear to necessitate screening for iron deficiency to ascertain the reliability of HbA1c in this population.11 Our observation is explained by the greater variance associated within the anaemia group. The significantly higher variance observed in the anaemia may be explained by the convenient homogenization of clinically heterogeneous anaemia entities in the anaemia group. Perhaps a prospective study that avoids this may report differently.
The significantly higher variance (23%) in the anaemia is explained by the heterogeneity of the subtypes within the anaemia group. The myriad of pathophysiologies (from variant haemoglobin affecting structure and function, and perhaps glycation rates of haemoglobin, to shortened erythrocyte lifespan due to intravascular and extravascular haemolysis) accounts for a less precise HbA1c reference range for the anaemia group. Separating the anaemia group into unique anaemia subtypes created less heterogeneity, reduced some within group variance and yielded a more precise references range for some anaemia subtypes.
The widened 95% CI of mean and reference ranges observed with mixed and sickle cell anaemia (95% CI of mean =4.82-5.59 and 4.84-6.26 respectively) may be attributable in part to the small number of participants in these subgroups (11 and 5 respectively, the normal curve is less robust in these circumstances [when n<30])). Furthermore, the marked variability in the type, severity, and the number of chronic morbidities and deficiencies causing mixed anaemia may be contributing. The imprecision of HbA1c observed with the sickle cell may be compounded by the unstable clinical course of sickle disease, marked by periodic crises with fluctuating [Hb] associated with intermittent or chronic haemolysis. These observations make the case for defining HbA1c reference ranges for each anaemia type.
A modest correction of anaemia (Δ [Hb] of +0.9g/dl, i.e <1g/dl) did not appear to cause a significant change in HbA1c levels. It is possible that higher increments in [Hb] may produce significant change in HbA1c (we predict in the direction of increment). A similar pattern was observed with anaemia of chronic disease and iron deficiency anaemia subtypes, where improvements in [Hb] of 0.6 and 1.3g/dl respectively did not cause a significant change in HbA1c. We propose that with anaemia of chronic disease, the change in [Hb] concentration was too modest to cause a significant change in HbA1c. The relative small size of participants (33) examined also makes type II statistical errors highly likely. We further propose that with anaemia of chronic disease, the myriad of functional cellular and system abnormalities (many, potentially affecting cellular homeostasis, especially acid-base balance and haemoglobin molecule covalent binding) associated with the primary disorder may impact on the potential for increase in HbA1c with increasing [Hb]. In view of the retrospective nature of the study, we could not ascertain the timelines of certain interventions and hence accurately determine the persistence of anaemia correction. Theoretically, a recent correction in [Hb] is less likely to impact on HbA1c. As alluded to above Kim et al (2010) evaluated for changes in HbA1c two months after correction of anaemia. Similar explanations are offered for the observation with iron deficiency anaemia. There were only 21 participants in the iron anaemia subgroup (i.e. <30, probable violation of a rule for use of parametric tests), making the parametric statistical tests less robust for the analysis. We did not study patterns with mixed, macrocytic and SCD, as each subtype had <7 (1,6,1) participants.
The study examined a large volume of data, eliminating as much as possible, potential extraneous factors in the relationship between [Hb] and HbA1c levels. However, the retrospective nature of the study made the control of other extraneous variables and certain patient attributes infeasible. It was also difficult to discern critical timelines and hence eliminate the potential impact of certain therapeutic interventions. Also, our exclusion of the younger population of patients (i.e. 16-20 years) does not necessarily indicate the result of the study may not be extended to this population of anaemia patients. In fact the similar human haemoglobin physiology in this group advises that the results may be extended to this younger population without concern. Due to the retrospective nature of the study, and in our attempt to increase inclusiveness, we allowed haemoglobin concentration and HbA1c assays done within a month of each other. In reality though, the majority (57%) had same day assays and even a greater majority (79%) had within same week assays. We recommend a larger scale prospective study with participants representative of all anaemia subtypes and ages so that the results can be extrapolated to the general population of anaemia patients.
Conclusion
The study emphasizes the need to exercise caution when applying HbA1c reference ranges to anaemic populations. It makes the case for defining HbA1c reference ranges and thus, therapeutic goals for each anaemia subtype. Redefining such reference ranges may increase the sensitivity of HbA1c in diagnosing diabetes in anaemic population if indeed the lower mean HbA1c (observed in this study) translates into significantly lower upper limits of references ranges (not observed in this study). Also, the realized reduced lower limits of reference range in this population will lead to appropriate clinical tolerance for lower HbA1c levels, with avoidance of inappropriate intervention for erroneous perception of over-enthusiastic control of diabetic hyperglycemia. We recommend that, absent risks factors for and symptoms relatable to diabetes, marginal elevations in HbA1c levels (i.e. HbA1c >6%) in anaemic patients should warrant confirmation of diagnosis using fasting blood glucose and 2HPPG or OGTT. The use of other surrogates of glycemic control, immune to the blur associated with haemoglobin type and concentration, may circumvent the problem associated with use of HbA1c in this special population. To this end, fructosamine and glycated albumin assays are currently being examined. 1,15
Metastatic carcinoma to the sinonasal tract is rare. We describe a patient with an aggressive follicular variant of papillary thyroid carcinoma who presented with an unusual metastasis to sphenoid sinus.
Case report
A 44 year old Hispanic woman presented at Queens Hospital Center in June 1988 with airway obstruction and was found to have a 10x12 cm firm mass in the left thyroid lobe, and palpable left supraclavicular node. She had no prior history of radiation, and no family of thyroid cancer. She underwent a total thyroidectomy with a modified radical neck dissection. Pathology revealed a follicular variant of papillary thyroid carcinoma: non-tall cell variant. Six of fifty (6/50) lymph nodes were positive. Post-surgery, patient received Iodine-131 ablation therapy (93 mCi) and was placed on thyroid hormone suppressive therapy. Non-stimulated thyrogen total body scan a week after therapy was negative. Thyrogen was not available at that time.
The patient was non-compliant with thyroxine and thyroid stimulating hormone (TSH) was often elevated (13-80 mlU/ml). However, the serum thyroglobulin remained less than 5.0 ng/ml and antithyroglobulin antibody was negative. A repeat total body scan (with 5 mCi I131) 6 months later and 4 years later with thyroxin withdrawal (TSH 36 mIU/ml and 48 mIU/ml respectively) was negative, and patient was continued on thyroxine suppression therapy.
Five years after initial presentation, the patient developed urinary retention and lower extremity weakness. A myelogram revealed block at T1-T2. Patient underwent laminectomy. Pathology revealed metastatic follicular variant of papillary thyroid carcinoma. Since iodine containing contrast was used during the myelogram, I131 iodine therapy was not given. External radiation of 2000 CGY to C7-T5 was administered.
A total body scan 8 weeks post laminectomy (when 24 hour urine iodine < 100 microgram/litre, and TSH was 38 mIU/ml after thyroid hormone withdrawal) was negative, the thyroglobulin level was 5 ng/ml and negative antithyroglobulin antibody (at that period of time, positron emission tomography (PET) scan was not an available option). For the next 2 years of follow up, the patient was maintained on thyroxin suppression therapy, this time with good compliance (TSH 0.1 mIU/ml, thyroglobulin less than 5 ng/ml and negative antithyroglobulin antibody). She did not show up for follow up lumbar computerised tomography (CT).
Seven years after the initial presentation, she complained of headache and double vision, and a three month history of amenorrhea. The thyroglobulin at this time was elevated (20 ng/ml). Chest X-ray was positive for two nodules in the right lung. Magnetic resonance imaging (MRI) revealed a soft tissue mass in the sphenoid sinus, eroding the basi-sphenoid and extending into the nasopharynx (Fig. 1 ABCD). The mass also eroded the sella floor displacing the pituitary gland upwards (arrows). Bone scan revealed focal abnormalities in the upper thoracic spine, ethmoid bones and base of the skull. At that period of time, PET scan was not an available option. Pituitary function testing revealed TSH 0.1 mIU/ml, free T4 level 1.2 mIU/ml. AM cortisol 5.3 mcg/dl, prolactin 182 ng/ml, ACTH 12 pg/ml, FSH 11.5 mIU/ml, LH 4.0 mIU/ml, and Estradiol 20 pg/ml.
Figure 1: A-T1 weighted midline sagittal MRI scan without contrast. B-T1 weighted midline sagittal MRI scan with contrast. C-T2 weighted axial MRI scan through the lesion. D-Axial CT scans without (on the left) and with (on the right) contrast. Note: The large destructive and enhancing lesion (*) in the sphenoid sinus associated with destruction of the basisphenoid, clivus and sellar floor. Note the normal pituitary gland (arrow) is displaced upwards out the sellaturcica.
Biopsy of the sphenoid sinus mass confirmed that it was metastatic papillary thyroid cancer, follicular variant. The tumour cell nuclear DNA was diploid and P53 and K167 were negative (Impat, NY). The patient was placed on hydrocortisone replacement and continued on thyroxine suppression therapy. Three months later the patient suffered a cardiorespiratory arrest and expired.
Discussion
Metastasis to the sphenoid sinus is rare from any tumour, and from papillary thyroid cancer it is extremely rare. An extensive world literature review revealed only 4 cases of spread to sphenoid sinus region from papillary thyroid cancer.1-4
Renal cell carcinoma is the most common tumour of paranasal sinus metastasis, 41.8%. The average age is 58 years, with slight predominance of males. The most common presentation was epistaxis, 31%. The most common causes of sphenoid metastasis are gastrointestinal and renal tumours5.
Von Eiselsberg et al. in 1893 described one case of metastasising thyroid carcinoma to sphenoid sinus.6 Harmer et al., 1899, reported a case of medullary thyroid carcinoma metastasis to sphenoid/ ethmoid sinus and nose. 7 Barrs et al. in 1979 reported a case of metastasis of follicular thyroid carcinoma to sphenoid sinus and sphenoid bone.8 Chang et al. in 1983 described a case of metastatic carcinoma of the thyroid to the sphenoid sinus.9 Renners et al. in 1992 reported one case of metastasis of follicular thyroid carcinoma to the paranasal sinuses, including the sphenoid sinus. 10Yamasoba et al. in 1994 reported a case with follicular thyroid carcinoma metastasising to sinonasal tract which also included sphenoid sinus.11 In the same year, Cumberworth et al. reported a case of metastasis of a thyroid follicular carcinoma to the sinonasal cavity which head CT showed sphenoid, ethmoid, frontal and maxillary sinuses. 12In 1997, Altman et al. described a case of follicular metastatic thyroid carcinoma to paranasal sinuses which included the sphenoid sinus. 13 The reported cases of thyroid cancer metastasis to sphenoid sinus are in table 1. Four cases were papillary thyroid carcinoma (included follicular variant of papillary thyroid carcinoma), six cases were follicular thyroid carcinoma, 1 case was medullary thyroid carcinoma and 1 case was unspecified thyroid carcinoma.
Table 1: Cases of thyroid metastases to the sphenoid sinus
Author
Age
Sex
Presenting symptoms
Histologic type
Present case
44
F
Headache, double vision and amenorrhea
Follicular variant papillary thyroid carcinoma
Mandronio (2011)
53
F
Blurring of vision of left eye
Papillary metastatic thyroid carcinoma
Nishijima (2010)
81
F
Epistaxis
Differentiated papillary thyroid carcinoma
Argibay Vasquez (2005)
53
F
Headache, paresthesia in the right eye region and left monocular diplopia
Differentiated carcinoma of thyroid, follicular variant of papillary cell
Altman (1997)
81
F
Progressive headache
Follicular thyroid carcinoma
Freeman (1996)
50
M
Facial pain, proptosis of the left globe and left horner’s syndrome
Metastatic papillary thyroid carcinoma
Yamatosoba (1994)
34
F
Hearing loss in right ear
Follicular thyroid carcinoma
Cumberworth (1994)
62
F
Right nasal blockage
Follicular carcinoma of the thyroid
Renner (1984)
61
F
Profuse right unilateral epistaxis
Follicular thyroid adenocarcinoma
Chang (1983)
50
F
Intermittent epistaxis, weight loss and pain in the right nasopharyngeal region
Follicular carcinoma with papillary foci
Barrs (1979)
54
F
Progressive loss of vision in the left eye
Follicular thyroid carcinoma
Harmer (1899)
44
F
Headache
Medullary thyroid carcinoma
von Eiselsberg (1893)
38
M
Chronic meningitis
Thyroid carcinoma
Pathologic lesions involving the sphenoid sinus include inflammatory disease, mucocele, chordoma, nasopharyngeal carcinoma, plasmacytoma, primary sphenoid sinus carcinoma, adenocystic carcinoma, pituitary adenoma, and giant cell granuloma. Benign disease often presents with a more gradual obstruction and disturbance of vision. This contrasts with the acute and progressive disturbances of vision in all cases reported with malignant lesions of the sphenoid sinus.14
Our patient presented with complaints of double vision for 6 months and headache. After imaging with MRI and given her previous history of metastatic thyroid cancer, the most likely diagnosis was metastases to the sphenoid sinus from the thyroid cancer, which was confirmed by tissue biopsy. Since this patient had evidence of bone metastasis, it is likely that the tumour first metastasised to the bone and then ruptured into the sphenoid sinus. The tumour appears to have eroded the sellar floor, extending into and displacing the pituitary gland, causing secondary hypoadrenalism.
In our patient, low thyroglobulin proved to be an unreliable marker because it was low when the patient had metastasis of the tumour in the spine. These tumours are more aggressive and today, PET scanning has proved more reliable in following them, a modality that was not available at the time for our patient. The possible explanations for negative total body scans in patients with metastatic differentiated thyroid cancer are a) technical limitations of the scan in detecting the tumour cells, and b) failure of the tumour tissue to trap iodine.
There are several unusual aspects in this patient’s presentation. Firstly, the initial presentation was unusual, since this tumour was very aggressive with rare sites of distant metastases. Perhaps the long periods of hypothyroidism when patient was noncompliant promoted the aggressive nature of this tumour. Secondly, the failure of known tumour markers, i.e. serum thyroglobulin and total body scan to identify these metastases. Thirdly, our patient’s tumour cell nuclear DNA was diploid. Investigations have shown that the DNA ploidy pattern as determined by flow cytometry is an important and independent prognostic variable.15-17 Fortunately, aggressive follicular variant papillary cancer of thyroid (non-tall cell type) is very uncommon.
Generally, total body scan negative with low stimulated thyroglobulin is an excellent prognostic sign. Our patient demonstrates that we need to remain vigilant for the unusual tumour especially when the initial presentation showed so much bulky disease. The need for additional tumour markers will help to identify aggressive well differentiated thyroid carcinoma cases.
Acknowledgement
Appreciation is extended to Ms. Deborah Goss and Mr. Timothy O’Mara, librarians, in helping with literature search and preparing the manuscript. No other financial sources or funding involved in the formation of manuscript. No potential financial conflicts of interest.
Diabetic patients with peripheral neuropathy are predisposed to foot injury. In Asian countries, a common culture among patients with peripheral neuropathy is to immerse their feet in hot water baths, with a belief that it will “improve circulation” and hence “cure the numbness”. We hereby report three cases of severe burn injuries of the feet presented to our hospital over a span of six months due to the above belief.
Case Report
The first patient was a 53-year old Malay gentleman with poorly controlled diabetes mellitus for six years, complicated with peripheral neuropathy, diabetic nephropathy and right eye cataract (latest HbA1C 8.1%), treated with oral anti-diabetic agents. He had a habit of using hot footbaths for numbness of both feet. Two weeks prior to presentation, due to increased feeling of numbness, he immersed his right foot into a self-prepared tub of hot water with added salt, followed by application of traditional sea cucumber gel. That evening, he noticed blistering of his right foot. Despite advice for admission, he chose to do the dressing as an outpatient in a local clinic. He presented two weeks later due to a worsening wound. At presentation, 4% full thickness burn of his right foot was noted, complicated by secondary infection (Figure 1). He underwent wound debridement, and subsequent split skin grafting. He had a prolonged hospitalization of five weeks due to secondary pseudomonas wound infection requiring parenteral antibiotics.
Figure 1. Right lower limb upon presentation to our hospital
The second patient was a 26-year old Indian gentleman with type I diabetes mellitus for nine years, complicated with diabetic nephropathy and peripheral neuropathy. His wife usually prepared hot water footbaths for him to improve his feet circulation. He developed 5% full thickness burn when he immersed his right foot into a pail of boiling water, not knowing that his wife had not added cold water into the footbath. He presented himself after two days and was hospitalized for two weeks. He recovered after wound debridement and split skin grafting.
The third patient was a 17-year old Chinese lady with poorly controlled type I diabetes mellitus for eight years, complicated with diabetic nephropathy (latest HbA1C 10.0%). She used hot water steam therapy with an aim to cure her recent onset of left foot drop, but was unaware of the temperature of the water. She developed blisters on her left foot, but only presented herself two weeks later when she developed left foot gas gangrene. She had a prolonged hospital stay of eight weeks with recurrent hospital acquired infections, including Methicillin-resistant Staphylcoccus aureus (MRSA). Despite multiple wound debridement, she required amputation of her left fifth toe (Figure 2).
Figure 2. Left lower limb post Ray amputation
Discussion
Peripheral neuropathy is a known complication of diabetes mellitus. More than 50% of patients who are over 60 years old have this complication.1, 2 Thermal injury to the feet in patients with neuropathy has been reported after walking barefoot on hot surfaces3 and after application of hot water bottles or heating pads during winter months.4, 5 The use of thermal footbath as a cause of burn injury is mostly due to patient-misuse or ignorance of correct usage.6, 7 In contrast, in Asian countries, a common culture among patients with peripheral neuropathy is to immerse their feet in self-prepared hot water without checking the water temperature,8 with a belief that it will “improve circulation” and hence “cure the numbness”. This practice has led to accidental burn injuries as described in our case reports.
There are a few reasons why patients with diabetic peripheral neuropathy end up with such a severe complication after the use of thermal footbath. Firstly, the temperature of the thermal bath may be underestimated. The time to develop full thickness burn reduces exponentially with just minimal increments in water temperature.9 Secondly, lack of pain despite the burn can prolong exposure to the heat source. In addition, concomitant peripheral vascular disease and endothelial function can limit vasodilatation to conduct heat away hence further aggravate the thermal insult.
Another important contributing factor of complicated wounds are the delays in seeking treatment as the result of lack of pain despite the burn injury. In a study done by Memmel et al on 1794 patients (of which 130 were diabetics) who presented with burn injuries, the majority of non-diabetic burn patients (63%) presented within 48 hours of injury, but only 40% of diabetic patients sought treatment within that time frame. Significantly more patients with diabetes presented after two weeks compared to those without diabetes. As burn injuries are highly susceptible to secondary infection, any delay in presentation further complicates and prolongs hospital stay.10,11 Not surprisingly, our two patients who presented two weeks after their burn injury had a prolonged and complicated hospital course compared to our second patient who presented soon after the burn injury. Increased susceptibility to infection and delayed wound healing from poor circulation contribute to prolonged recovery and poorer clinical outcomes in patients with diabetes mellitus, with some needing amputation as noted in our third patient.
As a healthcare provider we play a role in preventing this misfortune. Routine screening for the presence of peripheral neuropathy and vascular disease should be done during clinic visits to identify high-risk patients. Specific education regarding avoidance of thermal footbath and consequences of this highly preventable injury should be incorporated into standard diabetic foot care education. If patients choose to immerse their feet in hot water, temperature of the water should always be measured with a thermometer and immersion time should be limited. If a wound develops, they should present early to hospital for immediate treatment.
Conclusion
Thermal footbath for therapeutic purposes is commonly practiced in Asian culture. Our case reports highlight the serious consequences of this practice in diabetic patients with peripheral neuropathy. More public awareness and patient education is needed to prevent these injuries and to avoid the high cost of prolonged hospital stay and losses to the patient.
Population-based studies indicated that diabetes remains as a nationwide epidemic that continues to grow tremendously affecting 25.8 million people or 8.3% of the US population.1 This number is expected to reach 68 million or 25% of the population by 20302 as incidence of obesity is rising.3
The American Diabetes Association (ADA) recognizes diabetes education (DE) as an essential part of comprehensive care for patients with diabetes mellitus and recommends assessing self-management skills and knowledge at least annually in addition to participation in DE.4 With the objective of improving the quality of life and reducing the disease burden, the ADA and the U.S. Department of Health and Human Services through its Healthy People 2020 program have emphasized three key components for effective disease management planning: regular medical care, self-management education and ongoing diabetes support.5,6
The hallmark of preventing the chronic complications of diabetes lies in optimizing metabolic parameters such as glycaemic control, blood pressure, weight and lipid profile. Pharmacologic intervention can only do so much in achieving treatment goals. It should be complemented with appropriate DE emphasizing dietary control, physical activity and strict medication adherence.7,8 Adequate glycaemic control is clinically important because a percentile reduction in mean HbA1C is associated with a 21% reduction in diabetes-related death risk, 14% reduction in heart attacks and 37% reduction in microvascular complications.9
Diabetes self-management (DSM) education programs are valuable strategy for improving health behaviours which have significant impact on metabolic parameters.10 This is supported by chronic care model that is based on the notion that improving the health of patients with chronic diseases depends on a number of factors that include patients’ knowledge about their disease, daily practice of self-management techniques and healthy behaviors.11,12,13
A systematic review by Norris et al. has shown that DSM training confers positive effect on patients’ knowledge about diabetes, blood glucose monitoring, and importance of dietary practices and glycaemic control.14 In another retrospective observational study, evidence has suggested that participation in a multifactorial diabetes health education significantly improved glycaemic and lipid levels in the short term.10
Diabetes education/support group provides a comprehensive patient education, fosters a sense of community, and engages the patients to become active part of a team managing their diabetes. The diabetes support group at Queens Hospital Centre provides services to a diverse population from different socioeconomic backgrounds and is offered to any patients with diabetes. It is facilitated by certified diabetes nurse educators in the hospital and in the clinic. Patients meet once a month per session and are provided education in self-management of diabetes, education in medication, diet, lifestyle modifications, regular exercise, weight management and translation in their respective languages, if needed.
Few researches have been conducted comparing the efficacy of DE and combination of diabetes education and peer support group (DE+PS) in improving the metabolic parameters of patients with DM. In patients with DM, the primary objective of this study was to assess the clinical impact of DE and combined DE+PS group on metabolic parameters such as lowering HbA1C, reducing weight or BMI, controlling blood pressure, and improving lipid profile.
Methods
The study subjects were identified through retrospective review of electronic medical records of adult patients aged more than 18 years old with diabetes and being treated at the Diabetes Centre and/or Primary Care Clinic of Queens Hospital Centre, Jamaica, New York from January 01, 2007 to June 01, 2011. A total of 188 study subjects were selected and assigned to three groups: (1) control group (n=62), who received primary care only, (2) DE group (n=63), who received diabetes teaching from DM nurse educator in addition to primary care, and (3) DE+PS group (n=63), who received both diabetes education and attended at least 2 or more sessions of peer support group in addition to primary care. The subjects in control group, education group, education plus peer support group were matched on age, sex, weight and BMI. Considering the data availability, the duration of follow up measured in each group varied; the control group was followed up for 8 months, the DE group for 13 months and the DE+PS group for 19 months. The changes from mean baseline to the third month, sixth month and final follow up period were calculated for the following metabolic parameters: HbA1C, weight, BMI, SBP, TC, HDL-C, LDL-C and TG-C. T sample T-test was used to compare statistical differences in the mean changes in the metabolic parameters in each group from baseline to follow up period. All data management and statistical analyses were conducted with MiniTab version 14. A p-value of less than 0.05 is considered statistically significant.
Results
Among the 188 study subjects included in our study between ages 20 to 88 years with mean age of 60, the predominant gender was female (n=132, 70%). African American makes up the majority (n=74, 39%), followed by Asian (n=40, 21%), Caucasian (n=34, 18%), Hispanic (n=22, 12%) and Indian (n=18, 10%). Majority of our patients with DM have concurrent hypertension (91%), hyperlipidemia (90%), and obesity (47%). See Table 1 for baseline demographics.
Table 1. Baseline demographic characteristics of the study population
Control [C] N=62
Diabetes Education [DE] N=63
Diabetes Education + Peer support [DE+PS] N=63
Baseline Characteristics
Age range (years) [median]
32-76 [61]
20-88 [58]
26-86 [62]
Sex-male [N (%)]
22 (35)
20 (32)
14 (22)
Race
African American
31 (50)
26 (41)
17 (27)
White
11 (17)
23 (37)
0 (0)
Indian
18 (29)
0 (0)
0 (0)
Asian
1 (2)
10 (16)
29 (46)
Hispanic
1 (2)
4 (6)
17 (27)
Comorbidities [N (%)]
Hypertension#
54 (87)
59 (94)
58 (92)
Hyperlipidemia¥
56 (90)
61 (97)
53 (84)
Obesity*
29 (47)
29 (46)
31 (49)
Active cigarette smoker
6 (10)
5 (8)
1 (2)
# Hypertension is defined as mean systolic blood pressure > 140 mmHg and/or diastolic > 90 mmHg measured on two separate occasions. These patients have either hypertension diagnosed prior to or after diagnosis of DM. ¥ Hyperlipidemia is defined as LDL > 100 mg/dl in patients with diabetes and diagnosis hyperlipidemia could be before or after diagnosis of DM. * Obesity is defined as body mass index (BMI) of at least 30 kg/m2 or greater.
The group analysis showed that the DE group had a statistically significant decrease in mean HbA1C (mean change: -0.78%, p=0.013), TC (mean change: -16.89 mg/dL, p=0.01) and LDL-C (mean change: -11.75 mg/dL, p=0.04) from baseline to final follow up (see Table 2). The DE group had non-significant mean weight gain of 2.17 pounds and BMI of 0.52 kg/m2.
* Final follow up varies for the three groups. 8 months for control (C), 13 months for education (DE) group and 19 months for education plus peer support (DE+PS) group
Although DE+PS group were observed to have decreased in mean HbA1C (-0.48%), weight (-0.38 pounds), SBP (-3.24 mmHg), TC (-4.43 mg/dL) and TG-C (-12.89 mg/dL) and increased in HDL-C (+095 mg/dL), they were not statistically significant from initial to final follow up period. There were greater improvements in HbA1C and SBP from baseline to final follow up in DE+PS group compared to the control group. Only the control and DE+PS groups showed a decrease in weight from initial to final follow up.
Between the two intervention arms, the DE group exhibited greater reduction compared to DE+PS group in mean HbA1C (-0.78 vs. -0.48%), SBP (-3.78 vs. -3.24 mmHg), TC (-16.89 vs. -4.43 mg/dL), LDL-C (-11.75 vs. +0.08 mg/dL) and TG-C (-14.75 vs. -12.89 mg/dL).
Discussion
Our results suggested that among patients with DM, the subjects who participated in DE exhibited significant reduction in baseline HbA1C, TC and LDL-C compared to control. Furthermore, the significant impact of DE alone on optimizing control of HbA1C and LDL-C appeared to persist through time. In addition patients who received DE+PS also demonstrated moderate improvement in HbA1C, SBP, TC and TG-C and HDL-C even though they were not statistically significant on final follow up. It must be noted that the baseline mean HbA1Cs were higher in both interventions DE and DE+PS groups compared to control group and this may be associated with greater reduction in HbA1C in the intervention groups and may skew the finding. Our study results showed that DE group had greater percentage reduction in HbA1C (9%) compared to DE+PS group (5%) from baseline to the first follow up. The average change in HbA1C and LDL-C levels recorded in our study is similar to what has been reported in a previous study which showed significantly greater improvement in mean glycaemic levels and LDL-C levels in patients who participated in DE.10
However our findings are in stark contrast to a previous study that showed that DE+PS intervention has led to substantially greater weight reduction and improvement in HbA1C at second month post-intervention compared to education and control group.15 This difference may be accounted for by the effect of sample size and the duration of follow up. The DE+PS group in our study included twice the number of patients being sampled compared to previous study (63 patients vs. 32 patients), and longer duration of follow up (19 months vs. 4 months)15. These differences are significant as they can influence the data trend.
In general, all groups had improvement in HbA1C, TC, TG-C levels, and SBP (though not significant). Only control and DE+PS groups had weight reduction and DE group had weight increase. Although the DE+PS group had improvement in most of the metabolic parameters they were not statistically significant throughout the entire follow up period compared to DE group. This scenario might be attributed to retrospective nature of the study, possible non-compliance of patients to medications, differences in duration of follow up between groups, and limited number of patients sampled thus hindering the appreciation of potential significant effect. The statistically significant differences in baseline HB A1C among the three groups could also explain the differing magnitude of change from baseline; DE group had higher baseline HbA1C compared to control group (9.3 vs. 7.5%; p=0.00) allowing for a greater change from baseline value. Similarly in DE+PS group, baseline HbA1C was considered statistically significant compared to control group (8.3 vs. 7.5%, p=0.018).
A previous randomized controlled trial assessing the effect of peer support on patients with type 2 diabetes with a 2-year follow up demonstrated no significant differences in HbA1C (-0.08%, 95% CI -0.35% to 0.18%), SBP (-3.9 mmHg, -8.9 to 1.1 mmHg) and TC (-0.03 mmol/l, -0.28 to 0.22 mmol/l).16 It was suggested that the effect of DSM education on glycaemic control is greatest in the short-term and progressively attenuated over time and this may suggest that learned behaviour changes with time.17,18 However, the result of the present study showed a persistently significant beneficial effect on HbA1C and LDL-C from the earliest follow up until the final month for patients receiving DE alone.
Previous meta-analysis of randomized trials of DSM education programs by Norris and colleagues (2002) demonstrated the beneficial effect of DE with estimated effect on glycaemic control (HbA1C) at -0.76% (95% CI: 0.34,1.18) compared to control immediately after the intervention.17 However, the findings of the present study on the effect of peer education are in direct contrast with the results of the randomized trial using the Project Dulce model of peer-led education showing significant improvement from baseline to the tenth month of follow-up in HB A1C (-1.5%, p=0.01), TC (-7.2 mg/dl, p=0.04), HDL-C (+1.6 mg/dl, p=0.01) and LDL-C (-8.1 mg/dl, p=0.02).19 This could be accounted for the different baseline values of the metabolic parameters in the present study, thus creating a bias in the magnitude of change.
It has been suggested that the most effective peer support model includes both peer support and a structured educational program. The emphasis on peer support is based on the recognition that people living with chronic illness can share their knowledge and experiences to one another.20 It has been observed that participants in peer support groups were not interested in the topic of diabetes itself but on the effect and meaning of the disease on the lives of the patients.21
There are a number of limitations to be taken into consideration when interpreting the results of our study. Since our study is a retrospective review of medical records, the data collection was limited to availability of the required clinical data. Some parameters were not possible to obtain on a consistently uniform time frame. This resulted in varying mean duration for the 3 study groups (8 months for control group, 13 months for DE and 19 months DE+PS group). Because of unavailability of some of the clinical parameters at a specific time frame, there were variables missing on the earlier follow-ups. Our study also examined the effect of the intervention over a relatively short time. A longer-term study is necessary to determine if the intervention has lasting impact on improving the metabolic parameters, uplifting the quality of life and preventing morbidity and mortality from diabetes. The limited sample size could also be important factor that may influence the generalizability of the data. The differing baseline values in the metabolic parameters could have blunted the appreciation of possible significant improvement in the metabolic parameters in the DE+PS group. Other confounding factors that were not analysed in the present study and could have affected the results include the use of insulin regimen among the different groups, initiation of additional oral hypoglycemic agents, medication adherence by the patients and adjustment by physicians, and whether the patients were seen by endocrinologists or not.
The present study suggested that participation in DE may assist with optimizing HbA1C, TC and LDL-C. The DE group had improvement in glycaemic control and other metabolic parameters. The significant metabolic improvement gained from DE appeared to be sustained over time. However, participation in both DE+PS showed relative improvement but not significant as it is likely due to confounding different baseline metabolic parameter and duration being compared. Our findings underscore the importance of DE as part of the treatment plan for patients with DM. The addition of peer support group may or may not contribute to significant improvement of metabolic parameters.
Bortezomib is a reversible proteasome inhibitor, currently approved by US FDA for use in multiple myeloma and mantle cell lymphoma. It has been shown to cause new onset and exacerbation of underlying congestive cardiac failure (CHF) in some case reports. Although the exact mechanism of bortezomib induced congestive cardiac failure is unknown, studies have shown dysregulation of ubiquitin proteasome system (UPS) in human cardiac tissues in end stage heart failure1-3. Furthermore, a study in rats has shown reduced left ventricular contractility after bortezomib administration, which was attributed to reduced ATP synthesis in mitochondria of cardiac myocytes4. Our case demonstrates new onset severe reversible left ventricular systolic dysfunction after 4 cycles of bortezomib in a 58 year old female with multiple myeloma. It highlights the importance of monitoring cardiac function in patients receiving bortezomib.
Case Report
A 58 year old female with past medical history of well controlled hypertension presented with severe low back pain, anorexia and unintentional weight loss of around 20 pounds over a period of 3 months in medical clinic. On evaluation of her routine laboratory tests, she was found to have haemoglobin of 6.5 g/dl, haematocrit of 19.9%, white blood cell (WBC) count of 3.9 x 103/cc, red blood cell (RBC) count of 2.18 x 106/cc and platelet count of 1.52 x 105/cc. Her blood urea nitrogen and creatinine was 10 mg/dl and 0.7 mg/dl respectively and corrected calcium level was 10g/dl. On liver function test, her total protein was 12.4 g/dl and albumin level was 2.8 g/dl. X-ray of lumbosacral spine revealed a compression fracture at the level of T12and L2 vertebra. Bone survey confirmed diffuse osteopenia, severe collapse of the body of T12 and partial collapse of L2 and L3. Due to the presence of severe anaemia and compression fractures, multiple myeloma was suspected. Urine protein electrophoresis showed two monoclonal protein bands with concentration of 46.8% and 4.8% and urine immunofixation showed two intact monoclonal IgA-Kappa immunoglobulin bands. Beta-2 microglobulin level was 5.49. Bone marrow aspiration and biopsy confirmed the diagnosis of multiple myeloma. Patient was staged as IIIA according to Durie-Salmon staging system.
Subsequently, patient was planned to be treated with eight cycles of bortezomib and dexamethasone, with bortezomib being given on day 1, 4, 8 and 11 of each cycle at a dose of 1.3 mg/m2 body surface area. Prior to initiation of chemotherapy, she received radiotherapy to spine as well. However, after completing the fourth cycle of bortezomib/dexamethasone, she was admitted to the hospital with generalized weakness, nausea and vomiting. Chest X ray revealed possible right lower lobe infiltrate or effusion along with increased bronchovascular markings and she was treated with antibiotics for suspected community acquired pneumonia. However, an echocardiogram was obtained due to bilateral crackles on physical exam and increased bronchovascular markings on chest X ray, which revealed dilation of left ventricle with left ventricular ejection fraction of 30-35%, diffuse hypo kinesis of left ventricle, mild mitral and tricuspid regurgitation and diastolic dysfunction with abnormal relaxation(Tajik grade I). Left ventricular septal and posterior wall thickness was 0.8 cm. Infiltrative Cardiomyopathy in the setting of multiple myeloma was unlikely due to the absence of bi-atrial enlargement, pericardial effusion and thick bright myocardium on echocardiogram. Cardiology consultation was sought and their impression was new onset left ventricular dysfunction due to bortezomib therapy.
Patient did not receive any further cycles of chemotherapy due to cardiotoxicity and was on optimal medical management for heart failure with lisinopril, carvedilol and isosorbide dinitrate. An echocardiogram was repeated four months after discontinuation of bortezomib, which revealed normal left ventricular contractility with global left ventricular ejection fraction of 55% and trace mitral regurgitation.
Currently, at 2 year follow up, her echocardiogram shows global left ventricular ejection fraction of 65%, trace mitral and tricuspid regurgitation and diastolic dysfunction with abnormal relaxation(Tajik grade I).
Discussion and Review of Literature
Botezomib is a novel proteasome inhibitor which acts by inducing bcl-2 phosphorylation and cleavage, resulting in G2-M cell cycle phase arrest and apoptosis5. US Food and Drug Administration (FDA) have approved bortezomib for use in multiple myeloma and mantle cell lymphoma. The common adverse effects of bortezomib observed in clinical trials and post marketing surveillance include thrombocytopenia, neutropenia, hypotension, asthenia, peripheral neuropathy and nausea. US package insert for bortezomib states that acute development or exacerbation of congestive heart failure and new onset of decreased left ventricular ejection fraction have been reported, including reports in patients with no risk factors for decreased left ventricular ejection fraction and it is recommended to closely monitor patients with risk factor for, or existing heart disease.
The role of ubiquitin proteasome system (UPS) in heart failure has been studied extensively in recent years. Two studies by Hein et al and Weekes et al in 2003 have shown presence of increased amount of ubiquitinated proteins and substrates in cardiac tissues of heart failure patients, indicating reduced activity of UPS in end stage heart failure1-3. Another study has shown impaired proteasome activity in hypertrophic and dilated cardiomyopathy likely secondary to post translational modification of proteasome6.However, in early stage heart failure, there is increased activity of UPS, resulting in remodelling and high cardiac output2. Bortezomib, by inhibiting UPS, would lead to accumulation of ubiquitinated proteins in cardiac myocytes, similar to that seen in end stage heart failure. A study in rats exposed to bortezomib alone showed development of left ventricular systolic dysfunction by echocardiography and reduced synthesis of ATP was observed in the mitochondria of cardiac myocytes4. However, the exact mechanism of bortezomib induced systolic dysfunction in humans is not clear.
There have been a few reported cases of bortezomib induced congestive cardiac failure in literature (Table 1). The amount of bortezomib administered before development of symptoms of heart failure was 20.8 mg/m2 in four patients, 3 mg/m2 in one patient and 10.4mg/m2 in one patient. Three of them have received prior anthracycline based chemotherapy. Complete reversibility of heart failure after discontinuation of bortezomib was documented only in two cases by follow up echocardiograms and brain natriuretic peptide levels7, 8. The patient described in our index case had well controlled hypertension and no additional cardiac risk factors at baseline. She developed non-specific symptoms, including weakness, nausea and vomiting after the fourth cycle of chemotherapy and was admitted to the hospital for community acquired pneumonia. However, an echocardiogram was obtained due to pulmonary congestion, which uncovered the diagnosis of left ventricular systolic failure. The two echocardiograms obtained at a follow up of 4 months and 2 years showed gradual improvement in ejection fraction to 55% and 65% respectively from 15% after chemotherapy with bortezomib.
We did a review of major clinical trials of bortezomib in patients with multiple myeloma, Waldenstrom’s macroglobulinemia and plasma cell leukaemia (Table 2) to investigate the incidence of congestive cardiac failure reported after administration of bortezomib. In APEX trial, the incidence of congestive cardiac failure was 2% in both bortezomib and high dose dexamethasone group 11. In a study on melphalan refractory multiple myeloma by Hjorth et al, 3 cases of congestive cardiac failure was reported in bortezomib-dexamethasone group and 2 cases were reported in thalidomide-dexamethasone group12. Another study evaluating the safety of prolonged therapy with bortezomib by Berenson et al reported 1 case of cardiomegaly and 1 case of pulmonary edema13. However, further studies are needed to specifically evaluate the incidence of congestive cardiac failure with bortezomib therapy.
In summary, our case and review highlights the importance of maintaining a high level of suspicion for development of congestive cardiac failure after therapy with bortezomib. Given the widespread use of bortezomib and new generation proteasome inhibitors in multiple myeloma, there might be increased incidence of new onset and exacerbation of underlying congestive cardiac failure in future. Currently, there is no guideline for routine evaluation and monitoring of cardiac function in all patients during the course of bortezomib therapy. Furthermore, it is unclear whether the severity of congestive cardiac failure is proportional to the cumulative dosage of bortezomib administration and also, if there is any correlation between onsets of congestive cardiac failure with the timing of bortezomib therapy. Further studies are required in future to address these issues.
Table 1: Review of cases of bortezomib induced congestive cardiac failure reported so far.
Author
Age/sex
Prior cardiac history and risk factors
Baseline cardiac function
Number of Bortezomib containing cycles
Exposure to other cardiotoxic medications
Amount of Bortezomib received before onset of cardiac symptoms
Lowest EF** after Bortezomib administration
EF on follow up visits
Voortman et al7
53/M
36 pack years of smoking and COPD
Echo not available; NT-Pro BNP 1389 ng/l
4
Gemcitabine
3 mg/m2
10-15% on Echo after 4 cycles
45% on MUGA scan at 6 months
Orciuolo et al9
73/M
NK*
NK
6
1 Anthracycline containing regimen
20.8 mg/m2
EF 25%
NK
Orcioulo et al9
61/F
NK
NK
4
2 Anthracycline containing regimens
20.8 mg/m2
EF 20%
NK
Orciuolo et al9
80/F
NK
NK
4
1 prior non anthracycline chemotherapy regimen received
20.8 mg/m2
EF 35%
NK
Hasihanefioglu et al10
47/M
None
EF 70% and normal coronary angiogram
2
1 cycle of Vincristine, Doxorubicin and Dexamethasone
10.4 mg/m2
EF 10%
EF 20% at 6 month follow up
Bockorny et al8
56/F
Hypertension, well controlled
NK
4
None
20.8 mg/m2
EF 20-25%
EF 55-60%
INDEX CASE
58/F
Hypertension, well controlled
NK
4
None
20.8 mg/m2
EF 30-35%
EF 55% at 4 month and 65% at 2 year follow up.
*NK: Not Known; **EF: Ejection Fraction
Table 2: Review of cases of congestive cardiac failure reported in clinical trials with bortezomib in multiple myeloma, Waldenström’s Macroglobulinemia and plasma cell leukaemia.
Authors (ref)
Study
Study population
Significant Cardiac events (n)
Berenson, J.R. et al. 200513
Safety of prolonged therapy with bortezomib in relapsed or refractory multiple myeloma
Frontline chemotherapy with bortezomib-containing combinations improves response rate and survival in primary plasma cell leukaemia
29 patients with untreated PPCL
None reported
Hjorth, M. et al. 201212
Thal-Dex vs. Bort-Dex in refractory myeloma
131 patients with Melphalan refractory MM
2 cases of cardiac failure in Thal-Dex group and 3 in Bort-dex group
Jagannath, S. et al 200916
Bortezomib for Relapsed or Refractory Multiple Myeloma
54 patients with relapsed or refractory MM
None reported
Jagannath, S. et al 201017
Extended follow-up of Frontline Bortezomib ± Dexamethasone for MM
49 patients with untreated MM
None reported
Kobayashi, T. et al. 201018
Bortezomib plus dexamethasone for relapsed or treatment refractory multiple myeloma
88 patients with relapse/refractory MM
None reported
Mikhael, J.R. et al. 200919
High response rate to bortezomib with or without dexamethasone in patients with relapsed or refractory multiple myeloma
638 patients with refractory or relapsed MM
None reported
Richardson, P.G. et al. 200320
A Phase 2 Study of Bortezomib in Relapsed, Refractory Myeloma
202 patients with relapsed MM
None reported
Richardson, P.G. et al. 200511
Bortezomib or High-Dose Dexamethasone for Relapsed Multiple Myeloma(APEX trial)
669 patients with relapsed MM
Congestive cardiac failure in 2% of each arm.
Rosino, L. et al. 200721
Phase II PETHEMA Trial of Alternating Bortezomib and Dexamethasone As Induction Regimen Before Autologous Stem-Cell Transplantation in Younger Patients With Multiple Myeloma
40 patients with newly diagnosed MM
None reported
Sonneveld, P. et al. 201222
Bortezomib Induction and Maintenance Treatment in Patients With Newly Diagnosed Multiple Myeloma
827 patients with newly diagnosed MM
Cardiac Disorders in 5% of patient in VAD group vs. 8% of patients in PAD group.
Yuan, Z.G. et al. 201123
Different dose combinations of bortezomib and dexamethasone in the treatment of relapsed or refractory myeloma
168 patients with relapsed MM
None reported
Suvannasankha et al 200624
Weekly bortezomib/methylprednisolone in relapsed multiple myeloma
29 patients with relapsed multiple myeloma
1 case of congestive cardiac failure
Conclusion
CHF is an infrequent but serious adverse effect of bortezomib. Cardiac function should be closely monitored in patients receiving bortezomib, as case reports have shown that these patients might present with non-specific symptoms like weakness and fatigue. Further studies are required to establish the frequency and mode of monitoring of cardiac function during and after bortezomib therapy.
A 26 year old male was brought to Emergency department with history of altered sensorium of 1 day duration. He had a 2 week history of fever prior to admission. On examination, meningeal signs were present. Fundus examination showed evidence of papilloedema and a round pale yellow spot near the optic disc (Figure 1). CT scan of head did not reveal any abnormality.
Mantoux test and HIV ELISA were negative. CSF analysis showed:
Glucose – 40mg/dl; Protein: 2gm/l;
Cell Count: 1200cells/µl;
Cell Type: 80% lymphocytes;
CSF VDRL- negative;
CSF Grams stain, India ink staining and Ziehl Neelsen staining were unremarkable.
What is the Fundus finding?
Roth Spot
Cotton Wool Spot
Choroidal tubercle
A-V malformation
Discussion:
Correct answer: 3) Choroidal Tubercle.
Intraocular tuberculosis is a rare event and occurs in 1% of all diagnosed cases of tuberculosis.1 It occurs by haematogenous spread of mycobacterial organism. Choroidal tuberculosis is the most common initial manifestation of intraocular tuberculosis. They may be seen in 1.4% to 60% of patients with different forms of tuberculosis and are highly specific for tuberculosis. 2, 3
Choroidal tubercles may be unilateral or bilateral and appear as polymorphic yellowish lesions with discrete borders. They are of 2 types, solitary tubercle or granuloma (seen in chronic tuberculosis) and choroidal miliary tubercles (seen in acute miliary tuberculosis). Their size varies from 0.4 to 5 mm and may be associated with retinal vasculitis, panuveitis, choroiditis and neuroretinitis.
When they involve macula they present with visual loss and any delay in appropriate treatment results in irreversible visual loss. Peripherally situated tubercles are asymptomatic. Definitive diagnosis can be daunting due to difficulty in getting ocular samples for histological evaluation, however when available reveal features of granulomatous inflammation. Fundus angiography exhibits hypofluorescence in early stages and hyperfluorescence in later stages.
On treatment they heal by varying degrees of scar formation and marginal pigmentation.4 Untreated tubercles grow into large tumour like mass called tuberculoma.
Roth spots are retinal haemorrhages with a pale centre and are associated with bacterial endocarditis. Cotton wool spots appear as fluffy white patches on the retina and are associated with diabetes. A-V malformations are developmental vascular anomalies and appear as marked arterial and venous dilation associated with a tortuous pattern of vessels. They may have an associated bruit or chemosis of the eye.
The presence of ocular tuberculosis may be subtle. A high index of suspicion is required for its diagnosis. Delay in treatment or misdiagnosis may lead to irreversible visual loss.
Neuroleptic malignant syndrome (NMS) is a life-threatening neurologic disorder associated with the use of neuroleptic agents. NMS is typically characterized by a distinctive clinical syndrome of mental status change, muscle rigidity, fever, and autonomic instability. Atypical cases may present without muscle rigidity and/or hyperthermia. Association of infection in an atypical case can make the diagnosis challenging. We describe a case of NMS in a patient who presented with acute onset of altered mental status complicated with aspiration pneumonia.
Case Report
A 22-year-old female with a history of schizophrenia and seizure disorder presented with acute onset of altered mental status. Her home medications included haloperidol, clonazepam, olanzapine, trazodone, topiramate, benztropine, and trihexyphenadyl. The patient was found unarousable by her mother. She had multiple suicidal attempts in the past (overdosed on acetaminophen, drank cleaning detergent, and cut her wrist). She usually medicated herself without supervision. On examination, the patient was drowsy, afebrile (Temp 36.8oC/98.3oF), hypotensive (BP 85/64 mmHg), tachycardic (HR 105 bpm), and tachypnic (24/min). She was noted to be grunting with both inspiratory and expiratory stridor; therefore she was intubated for airway protection. Initial drug screen was negative for all substances of abuse. Acetaminophen and salicylate levels were undetectable. Head CT was unremarkable. Supportive care was provided as the patient was suspected to be overdosed with multiple medications. On the second day of admission the patient developed fever of 38.9oC/102oF. Chest xray and chest CT showed bilateral infiltrations (Fig.1), so empirical piperacillin/tazobactam and vancomycin were started for aspiration pneumonia. Sputum culture came back positive for Methicillin-resistant Staphylococcus aureus. However, the patient remained febrile with a temperature of 39.6 oC /103.2oF, despite appropriate antibiotic treatment. We suspected that there might be some other coexisting condition causing high fever. Serum creatine kinase (CK) was checked and found to be 8,105 U/L, up from 106 U/L on admission. We considered the diagnosis of NMS based on alteration of mental status, hyperthermia, autonomic instability, and elevated CK level, with the use of neuroleptic agents, although the patient did not have any muscle rigidity. The patient was started on dantrolene in addition to intravenous fluid and antibiotics. Shortly afterwards temperature and CK level started to trend down (Fig.2,3). Dantrolene was subsequently increased to maximum weight-based dose. Her mental status was gradually improved and returned to baseline. She became afebrile on day 10 of dantrolene treatment and serum CK went back to normal level after 2 weeks. Then bromocriptine was started orally and continued for 2 weeks.
Figure 1A: chest xray on admission
Figure 1B: chest xray on second day of admission
Figure 1C: chest CT on second day of admission
Figure 2. Temperature trend after starting treatment for NMS
Figure 3. CK trend after starting treatment for NMS
Atypical presentations of NMS can sometimes be difficult to diagnose, as in our patient who presented with altered mental status, fever, and coexisting infection, in the absence of muscle rigidity. We emphasize the importance of a high index of suspicion of NMS in patients using neuroleptic agents.
Discussion
NMS is an idiosyncratic drug reaction to antipsychotic medications and a potentially life threatening condition that occurs in an estimated 0.07% to 2.2% of patients treated with antipsychotics.1 Patients typically present with fever, rigidity, changes in mental status, and autonomic instability, often attributed to first generation antipsychotics, in particular after the start of medication or an increase in dosage.2 Atypical cases of NMS without muscle rigidity and/or hyperthermia have been reported, usually associated with atypical antipsychotic treatment. It has been hypothesized that atypical cases represent early or impending NMS; however pathogenesis remains unclear.3 Risk factors that have been established in case series and case-control studies include agitation, dehydration, acute medical illness, concomitant use of other psychotropic drugs, intramuscular injections and high doses of antipsychotic medications.4-6
Complications of NMS are often consequences of its symptoms. Pneumonia is the most common complication found in 13% of patient with NMS, likely due to altered mental status combined with difficulty swallowing that lead to aspiration.7 Renal failure is the second most common complication (8%), as a result of rhabdomyolysis and myoglobinuria. Other complications have been reported including myocardial infarction, disseminated intravascular coagulation, deep venous thrombosis, pulmonary embolism, hepatic failure, sepsis, and seizure.8 Mortality rate of NMS was around 20-30%.9-10 With early identification and treatment, mortality has significantly reduced to averages 10%.6
Withdrawal of the causative agent is the first step in the management of NMS. Supportive therapy, in particular, hydration, fever reduction, and careful monitoring, is the mainstay of management of NMS. In mild cases, supportive treatment alone may be sufficient.4 Adding specific therapies, such as dantrolene, bromocriptine, and benzodiazepine to supportive measures has been shown to reduce time to complete recovery, from a mean of 15 days with supportive care alone, to 9 days with dantrolene, and 10 days with bromocriptine.1 In severe cases, empirical trial of specific pharmacological agents should be started promptly. Electroconvulsive therapy is found to be effective when pharmacotherapy has failed.11
Sometimes NMS is difficult to identify in the presence of critical illnesses which can obscure the manifestation of NMS. As in our case, the patient presented with altered mental status, fever, and autonomic instability which could be simply explained by the presence of pneumonia and sepsis. However, due to lack of clinical response after appropriate antibiotic treatment, other coexisting condition was suspected. It is important to have a high index of suspicion for NMS in the setting of antipsychotic therapy. An absence of muscle rigidity should not exclude a diagnosis of NMS when the rest of the clinical picture points to this diagnosis. Elevated CK level helps support the diagnosis of NMS in patients with atypical presentation. Discontinuation of offending agent and supportive care should initiate promptly, and specific pharmacotherapy should be considered in severe cases. An early diagnosis is the key to successful treatment and patient outcome.
Conclusion
NMS is a rare but potentially life-threatening condition. Atypical presentation makes it more difficult to identify in patients with critical illnesses. Aspirated pneumonia is one of the common complications of NMS and sometimes can obscure signs and symptoms of NMS and delay diagnosis. High index of suspicion for NMS in patients taking antipsychotics is crucial. If not recognized or left untreated, NMD may be fatal.
Candida species is a leading cause of nosocomial infections and the most common fungal infection in intensive care units. Candida infection ranges from invasive candidal disease to blood stream infections (candidaemia). The incidence of Candida infection has been rising over the past two decades, particularly with the use of immunosuppressive drugs for cancer and HIV1,2,3 , and most of these infections occur in ICU settings.4 Candida infection is associated with high mortality and morbidity. Studies have shown that mortality attributable to candidaemia ranges from 5 to 71% depending on the study. 5.6.7Candidaemia is also associated with longer length of hospital stay and higher cost of care.
Early recognition of Candida BSI has been associated with improved outcome. Candida sepsis should be suspected in a patient who fails to improve and has multiple risk factors for invasive and bloodstream Candida infection. A variety of risk factors identified for candidaemia include previous use of antibiotics, sepsis, immunosupression, total parenteral nutrition, central venous line, surgery, malignancy and neutropaenia. Patients admitted to ICU are frequently colonised with Candida species. The role of colonisation in Candida blood stream infection and invasive candidal disease has always been debated. Few studies support the use of presumptive antifungal treatment in ICU based on colonisation and number of sites colonised by Candida. The NEMIS study has raised doubt about this approach of presumptive treatment. The Infectious Disease Society of America (IDSA) 2009 guidelines identify Candida colonisation as one of the risk factors for invasive candidiasis, but warn about the low positive predictive value of the level of Candida colonisation. 8 We conducted a retrospective cohort study in our medical ICU to identify risk factors for Candida blood stream infections including the role of Candida colonisation.
Hospital and Definitions:
This study was conducted at Interfaith Medical Center, Brooklyn, New York. It is a 280 bed community hospital with 13 medical ICU beds. A case of nosocomial Candida blood stream infection was defined as a growth of Candida Species in a blood culture drawn after 48 hours of admission. Cultures in our hospital are routinely done by the Bactec Method – aerobic and anaerobic cultures. Cultures are usually kept for 5 days at our facility and if yeast growth is identified, then species identification is done. In our ICU it is routine practice to do endotracheal culture and urine culture for all patients who are on mechanical ventilator supports and failing to improve. In patients who are not mechanically ventilated, it is routine practice to send sputum culture and nasal swabs to identify MRSA colonisation.
Study Design:
This study was a retrospective cohort study. We retrospectively reviewed all patients’ charts admitted to our medical ICU from 2000 to 2010 which stayed in the ICU for more than 7 days, irrespective of their diagnosis. Data were collected for demographics – age and sex. Data were also collected for risk factors for candidaemia – co-morbidities (HIV, cancer, COPD, diabetes mellitus, end-stage renal failure (ESRF)), presence or absence of sepsis, current or previous use of antibiotics, presence of central venous lines, steroid use during ICU stay, requirement of vasopressor support and use of total parenteral nutrition (TPN). Culture results for Candida including species identification were obtained for blood, urine and endotracheal aspirates.
Statistical Methods:
Patients were divided in two groups based on presence or absence of Candida BSI. Demographic data and risk factors were analysed using the chi square test to look at the difference between the two groups. Endotracheal aspirates and sputum cultures were combined to create a group with Candida respiratory tract colonisation. Binary logistic regression with forward likelihood ratio method was used to create models. Different models were generated for risk factors. Interactions between antibiotic use, steroid use, vasopressor support and sepsis were analysed in different models. Interactions between urine cultures and endotracheal aspirates/sputum cultures were also analysed by a different model. The model with the lowest Akaike information criterion (AIC) was chosen as the final model. The candidaemia risk score was calculated based on this final model to predict the risk of Candida BSI. Receiver operating curve (ROC) analysis was used to select the best cut-off value for the candidaemia risk score. Candida species in urine and endotracheal aspirates were compared with Candida species in blood culture using the kappa test. Data were analysed using SPSS statistical analysis software version 18.
Study Results:
A total of 1483 patients were included in the study. 56 patients (3.77%) had a blood culture positive for Candida species. Table 1 demonstrates demographic characteristics of the study population. There were no significant differences in the both groups for age, sex, diabetes mellitus, COPD, HIV, cancer and ESRF. As demonstrated in the table, 82.1% of patients in candidaemia groups recently used or were taking antibiotics as compared to 39.6% of patients in groups with no candidaemia. The P value was significant for this difference. Similarly, 71.4% of patients in the group with candidaemia had sepsis as compared to 30.6% in the other group with a P value of 0.000. Use of vasopressor (severe septic shock) was different between two groups – 23.2% and 10.1%, P value of 0.004. Steroid use, central lines and total parenteral nutrition use was higher in the candidaemia group as compared to the group without candidaemia. Similarly the rate of positive Candida cultures in urine and endotracheal aspirates was higher in the candidaemia group as compared to the group without.
Table 2 shows that 57.1% of Candida BSI were caused by C. Albicans, 30.4% by C. Glabrata and 12.5% by C. Parapsilosis. This incidence rate of species is similar to that found in other studies. Table 3 shows the two models with the lowest AIC value. The only difference between these two models was antibiotic use- previous or current use of antibiotics compared to current use of antibiotic in sepsis. Table 4 shows that when multifocal site positivity (urine and endotracheal culture) were used in the model, the AIC value increased significantly. This means that when multifocal sites were used in place of individual sites for the model, good amounts of information were lost and this model did not have good predictive value as compared to the model where individual sites are used for prediction of candidaemia. The model with lowest AIC was chosen as the final model. Binary logistic regression analysis with forward conditional analysis showed that only TPN, central venous line, previous or current antibiotic use, endotracheal aspirate culture positivity for Candida species and urine culture positive for Candida species were included in a statistical significant model. The final model had a P value of 0.000. Odds ratio with 95% confidence intervals and respective P values for all these risk factors are shown in Table 5. Age greater than 65 years, sex, sepsis or septic shock, co-morbidities and steroid use were not significant risk factors for candidaemia.
From this model, the candidaemia risk score calculated would be: Candidaemia risk score = 1.184 for previous or current antibiotic use + 0.639 for presence of central venous line + 1.186 for total parenteral nutrition + 0.760 for positive endotracheal culture for Candida + 1.255 for positive urine culture for Candida.
Table 6 shows the relationship between the Candida strain identified in endotracheal/sputum culture to that in blood culture. Similarly, Table 7 shows the relationship between the Candida strain identified in urine culture and that in blood culture. Strains identified in endotracheal aspirate culture had a very high value for the Kappa test and urine culture had a moderate value for agreement by the Kappa test. Thus, it can be inferred that Candida strain identified in blood culture was very similar to that identified in urine or endotracheal culture.
Table 1: Demographic characteristic of study population
Characteristic
Candidaemia (total 56) N (% of candidaemia)
No candidaemia (total 1427) N (% of no candidaemia)
Chi Square
Age >65 years
34 (60.7%)
676(47.40%)
0.06
Male sex
27 (48.2%)
694(48.6%)
0.530
Diabetes mellitus
22 (39.3%)
506(35.5%)
0.325
COPD
1(1.8%)
75(5.3%)
0.206
HIV
9 (16.1%)
253(17.7%)
0.458
Cancer
4(7.1%)
99(6.9%)
ESRF
11(19.6%)
251(17.6%)
0.401
Previous or current antibiotic use
46 (82.1%)
565(39.6%)
0.00
Sepsis
40(71.4%)
436(30.6%)
0.000
Vasopressor support (Septic shock)
13(23.2%)
144(10.1%)
0.004
Steroid use
27(48.2%)
431(30.2%)
0.004
Central line
30(53.6%)
267(18.7%)
0.000
Total parenteral nutrition
7(12.5%)
29(2.0%)
0.000
Candida in endotracheal aspirate/sputum culture
13(23.2%)
112(7.8%)
0.000
Candida in urine culture
34(60.7%)
262(18.4%)
0.000
Table 2: Candida strains responsible for Candida blood stream infection
Species in the blood culture
Number (%)
Candida Albicans
32(57.1%)
Candida Glabrata
17 (30.4%)
Candida Parapsilosis
7 (12.5%)
Table 3: Models with lowest two AIC
Variables
-2 log likelihood
AIC
Previous or current antibiotic use CVP line Total parenteral nutrition Endotracheal culture Urine culture
394.822
406.822
CVP line Total parenteral nutrition Endotracheal culture Urine culture Current antibiotic use in sepsis
395.730
407.73
Table 4: Model with 2 sites positive for Candida
Variables
-2 log likelihood
AIC
Sepsis CVP line Total parenteral nutrition Endotracheal and urine culture
407.920
417.92
Table 5: Odds ratio with 95% confidence interval for risk factors for candidaemia
Effect
Co efficient(β)
Odds ratio
95 % Confidence limit
P value
Lower
Upper
TPN
1.186
3.274
1.263
8.486
0.015
CVP line
0.639
1.895
1.032
3.478
0.039
Antibiotic Use
1.184
3.268
1.532
6.972
0.002
Endotracheal/ sputum culture
0.760
2.150
1.078
4.289
0.030
Urine
1.255
3.508
1.926
6.388
0.000
Table 6: Endotracheal aspirate culture in candidaemic patients
Endotracheal/Sputum Culture
Blood Culture
Kappa Test For Agreement
C. Albicans
C. Glabrata
C. Albicans
9
0
0.83
C.Glabrata
0
3
C. Tropicalis
0
1
Table 7: Urine cultures in candidaemic patients
Urine culture
Blood culture
Kappa Test for the agreement
C. Albicans
C. Glabrata
C. Tropicalis
C. Albicans
15
5
1
0.47
C. Glabrata
1
10
0
C. Krusei
1
1
0
Discussion
Candida is the most common nosocomial fungal infection in the ICU. Candidaemia accounts for approximately 5-8% of nosocomial BSI in the hospitals in the US.9,10,11 It accounts for approximately 50-75% of the cases of invasive fungal infection in the ICU12,13 and its rate varies from 0.2-1.73 per 1000 patient days.9,14,15 In a study done by Theoklis et al., candidaemia was associated with a mean 10.1 day increase in length of stay and a mean $39,331 increase in hospital charges.16 A study of 1,765 patients in Europe found that Candida colonisation was associated with increased hospital length of stay and increase in cost of care by 8000 EUR.17 ICU patients are at increased risk of infection because of their underlying illness requiring ICU care, immunosuppressant use, invasive or surgical procedures and nosocomial transfer of infections. A number of risk factors have been identified in different studies. In a matched case-control trial, previous use of antibiotic therapy, Candida isolated at other sites, haemodialysis and presence of a Hickman catheter were associated with increased risk of candidaemia.13 Similarly age of more than 65 years, steroid use, leucocytosis and prolonged ICU stays were risk factors for Candida BSI in 130 cases.18 Surgery, steroids, chemotherapy and neutropaenia with malignancy are the other identified risk factors.19
Candida BSI has a very high mortality rate. The attributable mortality varies from 5-71% in different studies.5,12,16,20 Even with treatment, there is high mortality as demonstrated in a study by Oude Lashof et al where out of 180 patients treated for candidaemia, 33% died during treatment and 55% completed treatment without complications.21 Risk factors for increased mortality in patients receiving antifungal treatment are delayed Candida antifungal treatment or inadequate dosing.22 Multivariate analysis of 157 patients with Candida BSI, APACHE II score, prior antibiotic treatment and delay in antifungal treatment were independent risk factors for mortality with odds ratio of 1.24, 4.05 and 2.09, respectively.23 Delayed treatment is also associated with increased fluconazole resistance as compared to early treatment and preventive treatment.24 Inadequate antifungal medication dose and retention of central venous catheters were also associated with increased mortality in a study of 245 Candida BSI, with adjusted odds ratios of 9.22 and 6.21, respectively.25,26
Candida albicans accounts for 38.8-79.4 % of the cases of Candida BSI. C. Glabrata is responsible for 20-25% of cases of candidaemia and C. tropicalis is responsible for less than 10% of cases of candidaemia in the US.9,20 ICU patients are frequently colonised with different Candida species. Candida colonisation can be from either endogenous or exogenous sources. Candida colonisation rates vary with the site- tracheal secretion (36%), throat swabs (27%), urine (25%) and stool (11%).27 Candida colonisation increases with the duration of the stay, use of urinary catheters and use of antibiotics. 28,29,30
The role of Candida colonisation in Candida BSI is frequently debated. Some studies have suggested that Candida colonisation of one or more anatomical sites are associated with increased risk of candidaemia.31,32,33,34 Typically, 84-94% of the patients developed candidaemia within a mean time of 5- 8 days after colonisation according to two studies.35,36 In another study, only 25.5% of colonised patients developed candidaemia.37 Similarity between strain identified in blood culture and that identified at various colonising sites was observed in one study.38 Candida colonisation by exogenously acquired species has also been implicated as a cause of candidaemia.39 In one study, 18-40% of cases of candidaemia were associated with clustering defined as “isolation of 2 or more strain with genotype that had more than 90% genetic relatedness in the same hospital within 90 days.” 40 Similar correlations for clusters are also noted for C. tropicalis candiduria41 and for C. Parapsilopsis.42 In a prospective study of 29 surgical ICU patients colonised with Candida, the APACHE II score, length of previous antibiotic therapy and intensity of Candida colonisation was associated with a significant risk of candidaemia. The Candida colonisation index calculated by non-blood body sites colonised by Candida over the total number of distinct sites tested for patients, was associated with a 100% positive and negative predictive value of candidaemia.29 Other studies do not support Candida colonisation as a risk factor for candidaemia. In a case-control study of trauma patients, only total parenteral nutrition was associated with an increased risk of candidaemia. Candida colonisation, steroid use, use of central venous catheters, APACHE II score, mechanical ventilation for more than 3 days, number and duration of antibiotics, haemodialysis, gastrointestinal perforation and number of units of blood transfused in first 24 hours of surgery. were not significant risk factors for candidaemia.43 NEMIS study found that in a surgical ICU, prior surgery, acute renal failure, total parenteral nutrition and triple lumen catheters were associated with increased risk of candidaemia; the relative risk for each risk factor being 7.3, 4.2, 3.6 and 5.4, respectively. Candida colonisation in urine, stool or both were not associated with increased risk of candidaemia.15
The effect of Candida colonisation of the respiratory tract on candidaemia and on mortality and morbidity is unclear. In a retrospective study of 639 patients, Candida respiratory tract colonisation was associated with increased hospital mortality (relative risk of 1.63) and increased length of stay (median increase of 21 days).30 In a study of 803 patients by Azoulay et al., respiratory tract colonisation was associated with prolonged ICU and hospital stays. These colonised patients were at increased risk of ventilator-associated Pseudomonas pneumonia, with an odds ratio of 2.22.44 However, in a postmortem study of 25 non-neutropaenic mechanically ventilated patients, 40% of the patients were colonised with Candida, but only 8% had Candida pneumonia.45,46 Jordi et al. found that out of 37 patients, definite or possible colonisation was found in 89% of patients and only 5% of cases were defined as Candida BSI.47.The effect of candiduria is also ill defined. Candida colonisation in urine has been implicated as a risk factor in certain studies. In a study done by Bross J et al., central lines, bladder catheters, 2 or more antibiotics, azotaemia, transfer from another hospital, diarrhoea and candiduria were significant risk factors for candidaemia. Candiduria had an odds ratio of 27 for development of candidaemia.48 Similar findings about candiduria were noted by Alvarez-Lerma et al.49
IDSA recommends starting empirical antifungal treatment for high risk neutropaenic patients who fail to improve on antibiotics after 4 days. Recommendation to start empirical antifungal therapy in low-risk neutropaenic patients and non-neutropaenic patients are not made by IDSA because of low risk of candidaemia.8 However, early detection of Candida BSI is vital because of increased mortality associated with delayed antifungal treatment and failure to remove central venous lines. Early detection of Candida BSI in a colonised patient can be facilitated by using a score based on the risk factors.50,51Similarly, b-D glucan assays can be used in patient colonised with Candida, to determine Candida BSI and need for antifungal treatment.52 Combined used of such risk factor identification systems and b-D glucan assays will help to detect candidaemia in earlier stages and will decrease mortality. Our study suggests that total parenteral nutrition, previous or current antibiotic use, central lines, candiduria and respiratory tract colonisation are risk factors for Candida BSI. With the help of our candidaemia risk score system, a score of more than 2 is associated with a higher risk of Candida BSI. This risk factor scoring system along with b-D glucan assays can be used to detect Candida BSI in earlier stages.
Conclusion:
Our study suggests that urine or respiratory tract colonisation is associated with an increased risk of Candida BSI, along with total parenteral nutrition, central venous lines and previous or current antibiotic use. We identified a scoring system which can be used along with a b-D glucan assay to detect candidaemia earlier.
It is well documented that many HBsAg-positive / HBeAg-negative patients show normal alanine aminotransferase (ALT) levels. However, two different scenarios have been proven to exist: inactive Hepatitis B Virus (HBV) carriers (previously defined as “healthy” HBV carriers) and patients with chronic hepatitis B (CHB) with transient virological and biochemical remission. These subsets of patients share HBsAg positivity and normal ALT levels; however, progression of disease, outcome, HBV DNA levels, severity of liver damage, requirement for liver biopsy and antiviral treatment significantly differ between the two patient populations.
Thus, among HBsAg-positive / HBeAg-negative subjects with normal liver biochemistry, it is important and sometimes difficult to distinguish the ‘true inactiveHBV carriers’ from patients with ‘activeCHB’ in whom phases of spontaneous remission have occurred. The former have a good prognosis with a low risk of complications, while the latter patient population have active liver disease with a high risk of progression to liver cirrhosis and/or hepatocellular carcinoma (HCC). Therefore, prolonged biochemical and virological follow-up are mandatory for diagnosis and decision to treat.
The term ‘chronic hepatitis B’ refers to a chronic necroinflammatory disease of the liver caused by persistent HBV infection 1. The term ‘necroinflammatory’ describes the presence of death of periportal hepatocytes (periportal necrosis) with or without disruption of the limiting plate by inflammatory cells, intralobular necrosis, portal or intralobular inflammation, and formation of bridges between vascular structures (the so-called bridging necrosis). Chronic hepatitis B can be subdivided into HbeAg positive and HBeAg-negative chronic hepatitis B 1,2. These two forms may have different natural histories and different response rates to antiviral treatment, although both may progress to more severe liver damage 3, such as, liver cirrhosis 4 or HCC 5.
The second subset is called the ‘inactive HBsAg carrier state’. It means a persistent HBV infection of the liver but without continual significant necroinflammatory disease. It is characterized by very low or undetectable serum HBV DNA levels and normal serum aminotransferases 1. It has been shown that histologically significant liver damage is rare in these patients, particularly when HBV DNA is lower than 2000 IU/ml, and thus a liver biopsy is not indicated in these subjects 4. Even among HBeAg-negative carriers with serum HBV DNA between 2000 and 20,000 IU/ml, histologically significant liver disease is also rare 6. Thus, these subjects should be followed up closely, but biopsy and treatment are not currently indicated.
As mentioned above, it is sometimes difficult to distinguish true inactive HBV carriers from patients with active HBeAg-negative CHB in whom phases of spontaneous remission may have occurred 1. The former patients have a good prognosis with a very low risk of complications, while the latter have active liver disease with a high risk of progression to advanced hepatic fibrosis, cirrhosis and subsequent complications such as decompensated cirrhosis and HCC 3-6. Thus, a minimum follow-up of 1 year with ALT levels every 3–4 months and periodical measurements of serum HBV DNA levels are required before classifying a patient as an inactive HBV carrier 1. ALT levels should remain consistently within the normal range, and HBV DNA should be below 2000 IU/ml 7. Thereafter, the inactive HBV carrier with undetectable or very low HBV DNA levels should be followed up with ALT determinations every 6 months after the first year and periodical measurement of HBV DNA levels 6 for the rest of their lifetime. This follow-up policy usually allows detection of fluctuations of activity in patients with true HBeAg-negative CHB 8.
It is important to underline that some inactive carriers may have HBV DNA levels greater than 2000 IU/ml (usually below 20,000 IU/ml), despite their persistently normal ALT levels 1,6,9. In these carriers the follow-up should be much more condensed, with ALT determinations every 3 months and HBV DNA measurements every 6–12 months for at least 3 years 1. After these 3 years, these patients should be followed up for life like all inactive chronic HBV carriers 6. After all, the inactive HBV carrier state confers a favourable long-term outcome with a very low risk of cirrhosis or HCC in the majority of patients 1,10,11. Patients with high baseline viremia levels have higher risk of subsequent reactivation. A liver biopsy should be recommended if ALT levels become abnormal and HBV DNA increases above 20,000 IU/ml. Non-invasive evaluation of liver fibrosis 12 may be useful, although these non-invasive tools, such as transient elastography, need further evaluation 6.
HBsAg clearance and seroconversion to anti-HBs antibody may occur spontaneously only in 1–3% of cases per year, usually after several years with persistently undetectable HBV DNA 7. On the other hand, progression to HBeAg-negative CHB may also occur 10.
Although the optimal definition of persistently normal ALT (PNALT) levels has not been established, the fluctuating nature of chronic HBV infection reasonably justifies serial ALT determinations. These should be done with a minimum of four to five tests 3–4 months apart within the first year of presentation, before determining whether an HBeAg-negative patient truly has PNALT. An initial follow-up of at least 1 year is supported by the finding of mild histological lesions in HBeAg-negative patients with true PNALT during the first year 6. The risk of developing abnormal ALT levels in HBeAg-negative patients with a normal baseline ALT have been reported to be higher during the first year (15–20%) and decline after 3 years of follow-up, therefore frequent monitoring during the first 1–3 years is critical 6,10.
Antiviral treatment of inactive HBsAg subjects is not indicated 1. Patients should be considered for treatment only when they have HBV DNA levels above 2000 IU/ml, serum ALT levels above the upper limit of normal and severity of liver damage assessed by liver biopsy showing moderate to severe active necroinflammation and/or at least moderate fibrosis 1,2.
The WHO estimated that 171 million people had diabetes in the year 2000 and predicted this number to increase to 366 million in the year 2030. Given the increasing prevalence of obesity it is likely that these figures provide an underestimate of future diabetes prevalence1. Peripheral diabetic neuropathy (PDN) may be present in 60 to 65% diabetic patients, with 11% patients of diabetic neuropathy complaining of pain. The management of this condition can be particularly challenging as these patients may not get good response to the medications used for the treatment and the medications used are associated with side effects which the patients may find difficult to tolerate.
Pathophysiology:
Pathophysiology of PDN is complex and incompletely understood. Both peripheral and central processes contribute to the chronic neuropathic pain in diabetes. Peripherally at the molecular level due to hyperglycaemia, glycosylated end products are generated, which deposit around the nerve fibres causing demyelination, axonal degeneration and reduction in nerve conduction velocity. Deposition of glycosylated end products around the capillary basement membrane causes basement membrane thickening and capillary endothelial damage, which in association with a hypercoaguable state causes peripheral arterial disease. The peripheral arterial disease leads to neuronal ischemia which worsens nerve damage. There also occurs depletion of NADPH by activation of NADPH oxidase causing increased oxidative stress and generation of oxidative free radicals which aggravate the nerve damage. Calcium and sodium channel dysfunction, changes in receptor expression are the other peripheral processes which cause further neuronal tissue injury. The nerve damage can cause neuronal hyperexcitability. Neurotropic factors are required for nerve regeneration. In diabetes there occurs a low level of both nerve growth factors and insulin-like growth factors resulting in impaired neuronal regeneration. This can lead to peripheral hyperexcitability. Central sensitization is cause by increased excitability at the synapse, which recruits several sub-threshold inputs and amplifies noxious and non-noxious stimuli. Loss of inhibitory interneurons, growth of non-damaged touch fibres into the territory of damaged pain pathways, increased concentration of neurotransmitters and wind up caused by NMDA receptors are responsible for central sensitization at the level of dorsal horn in the spinal cord2.
Clinical Presentation:
Chronic sensorimotor distal polyneuropathy is the most common type of diabetic neuropathy. Acute sensorimotor neuropathy is rare and is usually associated with diabetic ketoacidosis and acute neuritis caused by hyperglycaemia. Autonomic neuropathy is common and often under reported. It can affect cardiovascular, gastrointestinal and genitourinary system. The other presentations can be cranial neuropathies, thoraco-abdominal neuropathies or peripheral mononeuropathies involving median, ulnar, radial, femoral, lateral cutaneous nerve of the thigh or common peritoneal nerve.
The patients usually complain of one or more of the following symptoms. They can have a chronic continuous or intermittent pain described as burning, aching, crushing, cramping or gnawing pain. The pain can be associated with numbness. They can have brief abnormal stimulus evoked pain like allodynia or hyperalgesia. Some patients also complain of brief lancinating pains described as electrical or lightening pains which can be spontaneous or evoked. The symptoms typically start in the toes and feet and ascend in the lower limb over years and are worse at night. Diabetic distal polyneuropathy is typically described in glove and stocking distribution but the upper limb involvement is rare. On examination there can be paradoxically reduced sensation to light touch and pin prick in the area of pain. Examination can also show features of allodynia (pain caused by a stimulus that does not normally cause pain), hyperalgesia (pain of abnormal severity in response to a stimulus that normally produces pain), hyperpathia (painful reaction to a repetitive stimulus associated with increased threshold to pain), dysaesthesia (unpleasant abnormal sensation as numbness, pins and needles or burning), paraesthesia (abnormal sensation which is not unpleasant) or evoke electric shock like pains. There can be features of peripheral autonomic neuropathy including vasomotor changes like colour changes of feet which can be red, pale or cyanotic and temperature changes like warm or cold feet. With autonomic neuropathy there can also occur trophic changes which includes dry skin, callouses in pressure areas and abnormal hair and nail growth and sudomotor changes involving swollen feet with increased or decreased sweating. Mechanical allodynia is the most common type of allodynia, but there can be thermal allodynia described as cold or warmth allodynia. Patient often describes cold allodynia as the pain getting worse in cold weather and warmth allodynia can make the patient keep the effected limb cool by using fan or ice bags. There can be reduced joint position sense, reduced vibration sense, reduced temperature sensation and reduced ankle jerks.
Diagnosis:
The diagnosis of PDN can be made by clinical tests using pinprick, temperature and vibration perception (using a 128-Hz tuning fork). The feet should be examined for ulcers, calluses and deformities. Combinations of more than one test have >87% sensitivity in detecting PDN. Other forms of neuropathy, including chronic inflammatory demyelinating polyneuropathy, B12 deficiency, hypothyroidism and uraemia, occur more frequently in diabetes and should be ruled out. If required these patients should be referred to a neurologist for specialized examination and testing3.
Treatment Options:
US Food and Drug Administration has approved duloxetine in 2004 and pregabalin in 2005 for the treatment of painful DPN. Amitriptyline, nortriptyline and imipramine are not licenced for the treatment of neuropathic pain.
NICE clinical guidance on pharmacological management of neuropathic pain in adults in non-specialist settings as shown in Figure 1, recommends duloxetine as the first line treatment, which if contraindicated amitriptyline is suggested to be the first line. Second line treatment is amitriptyline or pregabalin. Pregabalin can be used alone or in combination with either amitriptyline or duloxetine, which if ineffective the patient should be referred to specialist pain services. While awaiting the referral tramadol can be started as the third line treatment. NICE also recommends that these patients should be reviewed to titrate the doses of the medication started, to assess the tolerability, adverse effects, pain reduction, improvement in daily activities, mood, quality of sleep and the overall improvement caused by the medication as reported by the patient4.
The Mayo clinic recommends5 the first tier of drugs for peripheral diabetic neuropathy are duloxetine, oxycodone CR, pregabalin and tricyclic antidepressants. The second tier of drugs iscarbamazepine, gabapentin, lamotrigine, tramadol and venlafaxine extended release. The topical agents suggested are capsaicin and lidocaine.
“Evidence Based guideline on treatment of Painful Diabetic Neuropathy”, was formulated by American Academy of Neurology; American Association of Neuromuscular and Electrodiagnostic Medicine; American Academy of Physical Medicine and Rehabilitation. A systematic review of the literature between time period 1960 to 2008 was carried out. The review included the articles which looked at the efficacy of a given treatment either pharmacological or non-pharmacological to reduce pain and improve physical function and quality of life (QOL) in patients with PDN. They subsequently recommended that Pregabalin is established as effective and should be offered for relief of PDN (Level A). Venlafaxine, duloxetine, amitriptyline, gabapentin, valproate, opioids (morphine sulfate, tramadol, and oxycodone controlled-release), and capsaicin are probably effective and should be considered for treatment of PDN (Level B). Other treatments have less robust evidence or the evidence is negative. Effective treatments for PDN are available, but many have side effects that limit their usefulness, and few studies have sufficient information on treatment effects on function and quality of life6.
Non-pharmacological techniques:
Letícia et al. following their literature review on therapies used for PDN concluded that for non-pharmacological techniques like acupuncture, reiki, photic stimulation, electromagnetic stimulation of neural electrical stimulation, laser therapy, there is a lack of consensus about their effectiveness and there is scarce knowledge about them. They suggested new researches, including treatments for a longer period of time, with dosimetry control, and representative samples are necessary to discover the actual importance of these therapies for pain relief7.Spinal Cord Stimulators have however been shown to be effective and safe in severe resistant cases8,9.
Pharmacological Therapies:
Pregabalin: Pregabalin is a gamma aminobutyric acid analogue which binds to α2δ subunit of calcium channels and modulates them. Its starting dose is 150 mg/day with a maximum dose of 600 mg/day. As 98% of the drug is excreted unchanged in the urine, its dose is reduced in patients with renal impairment (creatinine clearance below 60 ml/min)10. A Cochrane Database Systematic Review in 2009 showed that Pregabalin was effective at daily doses of 300 mg, 450 mg and 600 mg, but a daily dose of 150 mg was generally ineffective. The NNT for at least 50% pain relief for 600 mg daily pregabalin dose compared with placebo was 5.0 (4.0 to 6.6) for PDN11.
Duloxetine: Duloxetine reduces the reuptake of serotonin and noradrenaline at the level of spinal cord, thereby potentiating the descending inhibitory pain pathways to reduce pain. It is started at a dose of 30 mg/day and its dose can be increased to 120mg/day10. Sultan et al. in their systematic review found that pain relief achieved with daily dose of 60 mg Duloxetine was comparable with daily dose of 120 mg of Duloxetine. The number needed to treat (NNT) for at least 50% pain relief at 12 to 13 weeks with duloxetine 60 mg versus placebo was 5.8 as compared to NNT of 5.7 for daily dose of 120 mg of duloxetine. The side effects reported with Duloxetine were reduction in appetite, nausea, constipation and somnolence12. Systematic Reviews and cohort studies have shown that duloxetine provides overall savings in terms of better health outcomes and reduction in opioid use, in comparison to gabapentin and pregabalin, tricyclic antidepressants and venlafaxine, in pain caused by PDN13, 14.
Gabapetin: Gabapentin is structurally related to gamma-aminobutyric acid (GABA) but acts by binding to the alpha2-delta subunit of voltage-gated calcium channels and thereby reducing the transmission of neuronal signals. Gabapentin bioavailability is non-linear and it tends to decrease with increasing dose. Gabapentin is not bound to plasma proteins and has a high volume of distribution. It is eliminated unchanged by the kidneys so in elderly patients and in patients with impaired renal function, its dose must be reduced. Gabapentin can be started froma daily dose of 300 to 900 mg and the dose can be increased to a maximum daily dose of 3600 mg over 3 weeks period10. Gabapentin provides good pain relief in about one third of patients when taken for neuropathic pain. Adverse events are frequent, but most of them are tolerable15.
Tricyclic Antidepressants (TCA): Amitriptyline and nortriptyline are the commonly used TCA for PDN. They may be used if there is no benefit from pregabalin or gabapentin and duloxetine. They may be used alone or in combination with pregabalin or gabapentin. In small RCTs, amitriptyline has been found to relieve pain better than placebo in patients with diabetic neuropathy10. Amitriptyline is a tricyclic antidepressant with marked anticholinergic and sedative properties. It increases the synaptic concentration of noradrenaline and serotonin in the CNS by inhibiting their re-uptake by the pre-synaptic neuronal membrane. For neuropathic pain it is started at 10-25 mg orally once daily at bed time initially and increased according to response to a maximum of 150 mg/day.
TCA should be used with caution in patients with a history of epilepsy, cardiovascular disorders, deranged liver function, prostatic hypertrophy, history of urinary retention, blood dyscrasias, narrow-angle glaucoma or increased intra-ocular pressure. It’s other side-effects are agitation, confusion and postural hypotension in elderly patients10. Amitriptyline is the most studied TCA for DPN and has been compared with placebo, imipramine, and desipramine. Amitriptyline, when compared with placebo, reduced pain to a significant degree. Pain relief was evident as early as the second week of therapy, with greater pain relief noted at higher doses (at a mean dose of 90 mg). A decrease in pain was not associated with improvement in mood. A systematic review of the TCAs, including fewer than 200 patients, found no difference in efficacy between the agents16. Nortriptyline is associated with fewer adverse events than amitriptyline and therefore it should be preferred in elderly patients.
Opioids: The use of opioids in chronic neuropathic pain has been a topic of debate because of uncertainty about their effectiveness, the concerns about addiction problems, the loss of efficacy with their long term use due to development of tolerance with their long term use and the development of hyperalgesia associated with their use. Cochrane review of twenty-three trials of opiates was carried out. The short-term studies showed equivocal evidence, while the intermediate-term studies showed significant efficacy of opioids over placebo, in reducing the intensity of neuropathic pain. Adverse events of opioids were reported to be common but were not life threatening. The authors recommended the need for further randomized controlled trials to establish long-term efficacy, safety (including addiction potential) and effects on quality of life17. In RCT Tramadol/Acetaminophen combination was shown to be associated with significantly greater improvement than placebo (p < or = 0.05) in reducing pain intensity, sleep interference and several measures of quality of life and mood18. In another RCT, controlled release (CR) oxycodone was compared with placebo, CR oxycodone resulted in significantly lower mean daily pain, steady pain, brief pain, skin pain, total pain and disability. In this study the number needed to treat to obtain one patient with at least 50% pain relief is 2.619. Gabapentin and morphine combination in randomised controlled trial showed that the combination of the two drugs provided better analgesia at lower doses of each drug than either of the drugs used as a single agent20.
Capsaicin: Capsaicin is the active component of chilli peppers. Capsaicin works by releasing the pro-inflammatory mediators like substance P from the peripheral sensory nerve endings and thereby causes its depletion from the peripheral nerve. Pharmacological preparations of Capsaicin are available as 0.025% cream, 0.075% cream and 8% capsaicin patches10. Repeated application of a low dose (0.075%) cream, or a single application of a high dose (8%) patch has been shown to provide a degree of pain relief in some patients with painful neuropathy. Common side effect includes local skin irritation which causes burning and stinging. It is often mild and transient but sometimes severe and not tolerated by the patients leading to withdrawal from treatment. Capsaicin rarely causes systemic adverse effects. Capsaicin can be used either alone or in combination with other treatment to provide useful pain relief in individuals with neuropathic pain21.
5% Lidocaine medicated plasters: A recent systematic review showed that 5% Lidocaine medicated plaster causes pain relief comparable to pain relief caused by amitriptyline, capsaicin, gabapentin and pregabalin in treatment of painful diabetic peripheral neuropathy. Lidocaine plaster being a topical agent may be associated with lesser clinically significant adverse events than the side effects of systemic agents. The need for further studies has been recommended by the reviewer as limited number and size of studies were included in the systematic review 22.
Conclusion:
The American Academy of Neurology, Mayo Clinic and NICE have both developed guidelines for treatment of peripheral diabetic neuropathic pain. There are several peripheral and central pathological mechanisms leading to the development of this condition and no single drug is available to target all these pathological mechanisms. Therefore often a combination of drugs is required for their management. Despite using a combination of medicines, managing these cases can be challenging. At the same time there is limited evidence on combination therapy in diabetic neuropathy and much work is required in this area. While using opioids for this condition the controversies over the use of opioids in non-malignant pain should be kept in mind and the advantages and disadvantages of using them should be discussed with the patients. Opioids should only be started with patient’s consensus. The treatment should be modified from the guidelines on an individual basis to achieve the optimal pain relief.
Trigeminal Neuralgia (TN) is relatively rare in Multiple Sclerosis (MS) affecting approximately 2% of patients1. The severity of the pain is indistinguishable whether TN is an isolated impairment or is associated with MS. However, when associated with MS, TN is often bilateral, affecting younger patients and is more refractory to medical treatment 2.
Several pharmacological agents are reported to be effective in TN associated with MS. Topiramate3,4, gabapentin5 and lamotrigine6 were all reported to benefit patients with TN associated with MS in small uncontrolled trials. Several other drugs such as phenytoin, misoprostol and amitriptyline are routinely tried in patients with TN despite the lack of convincing evidence of their efficacy7.
In 2008, Both the American Academy of Neurology and the European Federation of Neurological Societies launched joint Task Force general guidelines for the management of TN. After systematic review of the literature the Task Force came to a series of evidence-based recommendations 8. Carbamazepine and oxcarbazepine had the strongest evidence of efficacy and were recommended as the first line treatment. An earlier Cochrane systematic review reached the same conclusion. 9.
Case report
A 62 year old female patient had been suffering from MS for about 20 years. The MS presented with trigeminal neuralgia from the outset and this was then followed by pyramidal lower limbs’ weakness and sphincteric dysfunction. The patient started to use a wheelchair 10 years ago but she became totally wheelchair dependent about 6 years later.
Trigeminal neuralgia remained active throughout the 20 years. Carbamazepine (300 mg daily) provided the patient with a satisfactory control of TN. Despite having occasional break through TN pain; the patient declined having higher doses of carbamazepine as excessive sedation was an unacceptable side effect.
Recently; the patient was admitted to hospital in two separate occasions complaining of increasing malaise and confusion. Plasma sodium levels were found to be low in both occasions (first presentation 118 mmol/l and second admission 114 mmol/l). Clinical evaluation confirmed Syndrome of Anti Diuretic Hormone Secretion (SIADH) as the cause of the hyponatremia and in the absence of any other explanation for the SIADH; carbamazepine was thought to be the main reason and was duly discontinued.
Unfortunately, TN attacks came back with vengeance. During the following 6 months, therapeutic trials using gabapentine, topiramate and amitriptyline failed to show any beneficial effect on either the severity or the frequency of the TN attacks. All three drugs were duly discontinued.
The patient was started on eslicrbazepine 400 mg on a single daily dose. This dose lead to almost complete eradication of the TN attacks. The control of TN and the plasma sodium levels remained stable a year following the initiation of the therapy.
Comments
Hyponatremia, defined as a sodium level < 135 mmol/l is a common side effect of carbamazepine and oxcarbazepine therapy. The incidence of hyponatremia secondary to carbamazepine therapy ranges between 4.8 and 40 % depending on the population studied10,11. In most cases, hyponatremia is asymptomatic and continuation of the carbamazepine use is possible whilst a close eye is kept on the plasma sodium level10. In rare occasions hyponatremia is symptomatic and discontinuation of carbamazepine is warranted. Administration of demeclocycline to normalise the sodium level was suggested by some authors12 However, the long term use of demeclocycline is associated with several complications and this approach is hardly a standard practice.
Clinicians often face a dilemma when carbamazepine is the only agent able to control a specific clinical problem. With many antiepileptics available, it is unusual to face such a problem in epileptic patients. Trigeminal neuralgia on the other hand can be extremely difficult to control and carbamazepine was found to have a unique ability to manage such unpleasant condition even before its antiepileptic effects were noticed on 196213.
Eslicarbazepine is promoted as an alternative to carbamazepine when side effects occurs on otherwise responsive patients to its favourable antiepileptic effects14. Hyponatremia is rare in eslacarbazepine users with only an incidence of less than 1% in the small populations studied15,16. Frequency of hyponatraemia increased with increasing eslicarbazepine dose. In patients with pre-existing renal disease leading to hyponatraemia, or in patients concomitantly treated with medicinal products which may themselves lead to hyponatraemia17.
Our patient showed the same favourable response to eslicarbazepine as she experienced with carbamazepine. However, hyponatremia did not occur with eslicarbazepine therapy. This enabled our patient to continue with pharmacological management and avoid surgical interventions.
With the exception of epilepsy, no reports are available commenting on the use of eslicarbazepine on the wide range of conditions that carbamazepine is traditionally used for such as mental health problems and neuropathic pain. When patients are well controlled on carbamazepine whatever the indication is, the occurrence of side effects such as hyponatremia is often managed by an automatic replacement with another agent. We feel that in such patients a therapeutic trial of eslicarbazepine might be appropriate especially if the control on carbamazepine was robust or if the benefits of carbamazepine therapy were clearly superior to other pharmacological agents potentially useful for the targeted clinical condition.
A 29 year-old woman had been well until 7 months previously when, after a viral syndrome, she developed palpitations, fatigue, and frequent episodes of light headedness and near syncope. On further questioning she notes exercise intolerance and dyspnea on exertion. She has stopped working as cashier. Her mother thinks she is having panic attacks and needs “something to calm her nerves.” ECG, echocardiogram, and endocrine evaluation are all normal. On physical examination, she displayed a postural heart rate increase of 35 beats per minute on standing, along with a 15mmHg fall in diastolic blood pressure.
Introduction
Disorders of the autonomic nervous system present unique challenges to the practicing clinician. These syndromes have a significant impact on quality of life, offer subtle diagnostic clues, and have a propensity to mimic other disease processes. For these reasons clinicians should have basic familiarity with the differentiating features of autonomic disorders. While much investigation has focused on Neurocardiogenic syncope, a distinct subgroup has emerged characterized by postural tachycardia and exercise intolerance. Postural orthostatic tachycardia syndrome (POTS) is the final common pathway of a heterogeneous group of underlying disorders that display similar clinical characteristics .1
The constellation of symptoms associated with POTS reflects underlying dysautonomia including palpitations, exercise intolerance, fatigue, lightheadedness, tremor, headache, nausea, near syncope, and syncope. Foremost amongst these is orthostatic intolerance. Besides being limited in the functional activities of daily living, increased sleep disturbances including excess daytime sleepiness, fatigue are also common and have been associated poor health-related quality of life. 2 We aim to discuss the clinical presentation, classification, evaluation and management of POTS.
Diagnosis
The current diagnostic criteria for POTS is the presence of orthostatic intolerance symptoms associated with a sustained heart rate increase of 30 beat per minute (bpm) or absolute rate exceeding 120 bpm within the first 10 minutes of standing or upright tilt in the absence of other chronic debilitating disorders, prolonged bed rest, or medications that impair vascular or autonomic tone. 3 Most patients will have orthostatic symptoms in the absence of orthostatic hypotension (a fall in BP >20/10 mmHg). A grading system for POTS has been developed (Table 1). This system focuses on the functional severity of orthostatic intolerance in a way similar to the NYHA heart classification.
The physical exam should be methodical and directed. Supine, sitting, and immediate standing heart rate and blood pressure should be recorded. Orthostatic tachycardia and acrocyanosis of the lower extremities may be the only physical signs in these patients. Standing and supine blood pressure measurements and baseline electrocardiogram are generally recommended. In patients with physical examination or clinical history suggestive of cardiovascular abnormalities, other diagnostic test including echocardiogram , stress test or coronary angiography may be indicated before a tilt test. In addition, other possible clinical disorders with similar clinical manifestations must be kept in mind and ruled out. Upright tilt test remains the diagnostic test of choice for POTS and other autonomic disorders. Implantable loop recorders, electrophysiological tests and Holter monitoring are not helpful in the evaluation of these disorders. Supine and upright serum catecholamine levels should be obtained if hyperadrenergic type is suspected. In the presence of gastrointestinal symptoms bowel motility studies may reflect the degree of involvement and help to tailor therapy. 4
Table 1 : The Grading of Orthostatic Intolerance*
Grade 0 Normal orthostatic tolerance
Grade 1 Orthostatic symptoms are infrequent or occur only under conditions of increased orthostatic stress Subject is able to stand >15 minutes on most occasions Subject typically has unrestricted activities of daily living
Grade II Orthostatic symptoms are frequent, developing at least once a week Orthostatic symptoms commonly develop with orthostatic stress Subject is able to stand >5 minutes on most occasions Some limitation in activities of daily living is typical
Grade III Orthostatic symptoms develop on most occasions and are regularly unmasked by orthostatic stresses Subject is able to stand for >1 minute on most occasions Patient is seriously incapacitated, being bed or wheelchair bound because of orthostatic intolerance Syncope / presyncope is common if patient attempts to stand
*Symptoms may vary with time and state of hydration and circumstances. Orthostatic stresses include prolonged standing, a meal, exertion, and head stress.
Classification and Clinical Features
POTS is a heterogeneous group of disorders resulting in a common clinical scenario. This syndrome is classified as being either primary or secondary. Primary POTS is idiopathic and not associated with other disease processes. Secondary POTS occurs in association with a known disease or disorder. Subtype classification affects management and is therefore essential.1,4 (Figure 1)
Partial dysautonomic (PD) POTS (also referred to as Neuropathic POTS) is the predominant form.1, 2 This is a mild peripheral autonomic neuropathy, characterized by inadequate peripheral vasculature constriction in the face of orthostatic challenge. Female predominance is seen with 5:1 female to male ratio. Presentation is commonly abrupt onset of symptoms after a febrile viral illness, pregnancy, immunization, sepsis, surgery, or trauma. The etiology of PD type POTS is postulated to be an autoimmune molecular antigen mimicry type pathogenesis. Serum autoantibodies to peripheral autonomic ganglion alpha-3 acetylcholine receptors may be positive in 10-15% of the cases. Anhidrosis of lower extremities is seen in more than 50% of these patients at quantitative sudomotor axon reflex testingDependent blood pooling when upright is greater than normal and heart rate and contractility increase as normal compensatory physiologic mechanisms to maintain cerebral perfusion. This autoregulatory response may initially be fully compensatory; however, peripheral venous pooling may increase with time and exceed this compensatory effect. Patients with PD type POTS become dependent on the skeletal muscle pump to augment their autoregulatory physiology. Ultimately, venous blood pooling increases beyond the body’s total ability to compensate and adequate blood pressure maintenance fails.
“Developmental” partial dysautonomic POTS is an adolescent subtype.6 The mean age of onset is 14 years. The clinical scenario is that of orthostatic intolerance following a period of very rapid growth. Symptoms are progressive and peak at a mean age of 16 years. Orthostatic intolerance may be severe, including severe headaches, and can be functionally disabling. Following their peak symptoms will slowly improve and resolve into young adulthood (19-24 years). Roughly 80% of patients with developmental PD POTS will experience complete resolution of symptoms. The etiology of this subtype is unclear and appears to be a transient period of autonomic imbalance occurring in rapidly growing adolescents.
Hyperadrenergic POTS is less common than the PD type ,7This form is characterized by a gradual onset with slowly progressive symptoms. Patients report experiencing tremor, anxiety, and cold clammy extremities with upright posture. 7 Many patients note increased urine output when upright. True migraine headaches may be seen in over half of patients.7 Gastrointestinal symptoms in the form of recurrent diarrhea were seen in 30% of the patients. In contrast to PD type of POTS, the hyperadrenergic form demonstrates elevated serum catecholamine levels with serum norepinephrine levels >600ng/ml. This may be a familial syndrome in some instances determined by a careful history. The etiology of hyperadrenergic POTS is felt to be genetic with a single point mutation resulting in a dysfunctional norepinephrine reuptake transporter protein present in the intrasynaptic cleft. The result is excessive norepinephrine serum spillover with sympathetic stimulation resulting in a relative hyperadrenergic state appearing similar to pheochromocytoma. 8
A connective tissue disorder has been an increasingly recognized etiology of secondary POTS 9 . Joint hypermobility syndrome (JHS) is an inherited condition characterized by joint hypermobility, connective tissue fragility, and soft velvety skin with variable hyperextensibility. The condition is associated with ecchymotic predisposition, premature varicose veins, diffuse muscle and joint pain, and orthostatic acrocyanosis. The etiology of POTS in JHS patients is thought to be due to abnormal vascular (venous) elastic connective tissue. During orthostatic stress and increased hydrostatic pressure these patients exhibit increased vessel distensibility and orthostatic intolerance. Excessive peripheral venous pooling and compensatory tachycardia follows. Up to 70% of JHS patients suffer from some degree of orthostatic intolerance. It is observed that adolescent PD POTS patients have features similar to JHS patients and further studies may determine the significance of this potential relationship 10.
Secondary POTS refers to a group of conditions which result in peripheral autonomic denervation with sparing of cardiac innervation. Most commonly, secondary POTS is associated with diabetes mellitus. Less commonly, this form may occur with heavy metal intoxication, multiple sclerosis, parkinsonism and chemotherapy, especially vinca alkaloids.11,12
Severe autonomic nervous system disorders may present as POTS. These may include pure autonomic failure or multiple system atrophy. Paraneoplastic syndrome associated with adenocarcinoma of the lung, breast, ovary, or pancreas may also present as POTS. These tumors produce auto-antibodies targeting the acetylcholine receptors in the autonomic ganglia in a manner similar to post-viral syndromes.
Figure 1. Subtypes of postural orthostatic tachycardia syndrome
Evaluation and Management
Treatment is generally individualized to each patient. Confounding pharmacology should be identified and stopped if possible (Table 2). The presence of secondary POTS should be considered. Underlying diagnoses causing or augmenting POTS should be identified and treated appropriately. Deconditioning is frequent seen in POTS patient and a deliberate aerobic reconditioning program should be a component of the treatment plan. 13 This is encouraged to begin promptly working up to a goal of 20-30 minutes of activity at least 3 times a week. Resistance training of the lower extremities is helpful to increase the efficacy of the skeletal muscle pump. Salt and water ingestion are the most common employed non-pharmacolgical therapeutic intervention for POTS. Although, the intravenous saline infusion has been associated with reduction in standing tachycardia, the effect on this intervention on the symptom reduction remains unknown 14 . In general, since low blood volume may exacerbate symptoms patients are encouraged to have liberal salt and water intake. Excluding hyperadrenergic POTS, daily fluid and sodium intake should be greater than 2 liters and 3-5 grams.
Table 2: Pharmacologic Agents That May Cause or Worsen Orthostatic Intolerance
The goal of pharmacotherapy in the treatment of POTS is to ameliorate the symptoms of POTS and thus maintain the functional capacity. Currently no drug is US FDA approved for the treatment of POTS. All pharmacology is inherently off-label. (Table 3).
Majority of the evidence for the use of different pharmacological agents in the management of POTS is based on some small randomised , observational and retrospective single center studies. In clinical practice most patients are treated with a single agent and second medication from different class with a different mechanism of action is added in case of treatment failure. Resistant cases are often treated with polypharmacy.
Fludrocortisone, a potent mineralocorticoid resulting in sodium retention, augmented fluid volume, and sensitized peripheral alpha adrenergic receptors. The effects are more pronounced in the younger population. Starting dose is 0.1-0.2 mg daily with a maximum dose of 0.4 mg. Common side effects include electrolyte imbalance and hypertension. In a study of 11 female POTS patients, fludricortisone alone or in combination with bisoprolol was associated with improvement in symptoms 15. Midodrine is an alpha -1 adrenoreceptor agonist and causes both arterial and venous vasoconstriction. It is commonly used as add on therapy and with starting dose at 5 mg orally three times a day. In our clinical experience we advise patients to take their first dose of midodrine 15 minutes prior to getting out of bed. An additional 5mg dose can be used for breakthrough symptoms. Midodrine is usually well tolerated with the most common complaints being nausea, “goose bumps,” and scalp pruritus. In a small study of 6 patients with POTS acute combination therapy of midodrine (10mg) with octreotide (0.9mcg/kg) was significantly associated with reduction in upright tachycardia and improved standing time.In another study of 53 children with POTS, midodrine was significantly associated with both higher clinical cure rate and reduced recurrence rate as compared to children treated with metoprolol or conventional therapy.
Patients may continue to be symptomatic despite dual-therapy as outlined above. In this population we add a serotonin reuptake inhibitor (SSRI) or norepinephrine reuptake inhibitor (SNRI). SSRI therapy has been found to be helpful in the prevention of neurocardiogenic syncope. However, SNRI therapy is more useful in the treatment of POTS. Usually, we use bupropion XL beginning with 150 mg orally daily titratable to 300 mg daily if necessary.
The most effective SSRI therapies combine serotonin and norepinephrine reuptake inhibition (venlafaxine and duloxetine). The agents are usually well tolerated with the most common side effects being gastrointestinal upset, tremor, sleep disturbance, and less commonly agitation and sexual dysfunction. Bupropion and SSRI therapy can be combined to achieve a similar effect.
Pyridostigmine is an acetylcholinesterase inhibitor that facilitates sympathetic and parasympathetic ganglionic neural transmission. In our single center experience of 203 patients of POTS treated with pyridostigmine; improved symptoms of orthostatic intolerance were seen in 88 of 203 (43%) of total patients or 88 of 172 (51%) who were able to tolerate the drug. Fatigue (55%), palpitations (60%), presyncope (60%), and syncope (48%) were the most common symptoms that improved with pyridostigmine. Further, symptom reduction correlated with a statistically significant improvement in upright HR and diastolic blood pressure after treatment with pyridostigmine as compared to their baseline hemodynamic parameters (standing HR 94 ± 19 vs 82 ± 16, P < 0.003, standing diastolic blood pressure 71 ± 11 vs 74 ± 12, P < 0.02). Gastrointestinal problems were the most common adverse effects (n = 39, 19%) seen in our study. 18
Severely affected and refractory patients may benefit from erythropoietin (EPO) therapy. EPO increases red cell mass, central blood volume and augments response of blood vessels to the angiotensin-II and thus causes vasoconstriction. These effects are quite useful in the treatment of orthostatic disorders. Prior to initiation of EPO therapy obtain a complete blood count (CBC), total iron binding capacity, serum iron and ferritin levels. Hematocrit (HCT) levels must be monitored and should remain less than 50 on EPO. The starting dose is 10,000 units via subcutaneous injection once weekly. There is a 4-6 week delay between a given dose and the full clinical effect. The hematogenic and hemodynamic affects are independent but may occur simultaneously. A goal HCT of low to mid 40 will often result in optimum hemodynamic augmentation. Monitoring during EPO therapy should include monthly CBC to document HCT less than 50. EPO therapy may infrequently result in a “serum sickness” type reaction characterized by nausea, fever, chills, and general malaise. In another study of 39 patients (age 33 ± 12, 37 females) with resistant form of POTS, we reported sustained improvement in twenty-seven (71%) patients at mean follow-up of six month with EPO therapy. Eighty (21%) failed to respond to therapy while as 3 (8%) improved with therapy at 3months. Also, erythropoietin significantly improved sitting diastolic blood pressure but had no effect on other hemodynamic parameters.19 We reserve EPO therapy for patients that are refractory or intolerant of other forms of treatment because of its considerable expense and subcutaneous route of administration.
Beta blocker therapy such as metoprolol tartrate may be beneficial in adolescent type POTS patients. In a single center retrospective study of 121 patients with possible POTS, written survey at follow-up were used to evaluate response to therapy with beta-blockers and midodrine. 47 adolescents responded to survey (Walker Functional Disability Inventory Survey) and reported improvement with a β-blocker (100% vs 62%, P = 0.016) and more attributed their progress to medication (63.6% vs 36.4%, P = 0.011) than did those treated with midodrine 20.In addition, beta-blocker therapy was associated with improved quality of life. Great caution should be taken in using beta-blocker therapy in a rare form of hyperadrenergic POTS secondary to the mast cell activation disorders. Octreotide is a somatostatin analogue with potent vasoconstrictive effects and is useful in the treatment of orthostatic disorders. In patients with resistant POTS, octreotide may be a useful as add on therapy. It is administered by subcutaneous injection 2-3 times daily. The stating dose is 50 ug and may be titrated up to 100-200 ug three times daily.
Agents that block the release or effect of norepinephrine (noradrenaline) are very effective for hyperadrenergic type POTS patients. We use clonidine starting at 0.1 mg orally twice daily and titrating up as needed. The patch form may be preferable to some patients and has the added benefit of providing a steady state drug release for one week. Labetalol, an alpha and beta receptor blocker, is also useful in this group of patients. Dosages of 100-400 mg orally twice daily are used. Methyldopa may have a role in highly selected patients with POTS. Symptom control may be improved with both the SSRI and SNRI classes of medications.
Inappropriate sinus tachycardia (IST) is an important confounding finding in suspected POTS patients. This syndrome is similar to hyperadrenergic type POTS. The clinical presentation may be similar with IST being more common in females. These disease states display and exaggerated response to isoproterenol infusion. It has been postulated that they may represent different states of the same pathologic process. A greater degree of orthostatic change in heart rate is seen in POTS patients. The supine rate rarely exceeds 100 bmp (IST will often be >100). Postural changes in serum norepinephrine levels are much more pronounced in POTS patients. It is important to differentiate POTS and IST. Radiofrequency ablation of the sinus node will rarely benefit hyperadrenergic POTS patients and will make PD POTS patients markedly worse.
Treatment of secondary POTS should focus primarily on the underlying disorder to the greatest extent possible. Diabetes mellitus or JHS related POTS are treated as PD POTS. Secondary POTS due to sarcoidosis or amyloidosis may benefit from steroid therapy. Secondary POTS that is paraneoplastic may completely resolve with treatment of the underlying malignancy but may also respond to pyridostigmine.
Patients suffering from POTS have a disease that affects many aspects of their life. They are often unable to take advantage of meaningful employment or education opportunities. The pervasive life change experienced often results in significant psychosocial disruption as they may be excluded from social norms and certain environments. Frequently, patients require psychologists, social workers, and lawyers to address these aspects of living with POTS. The treating physician is a prominent and central figure who is a beacon of hope for this population. A positive, caring, and nurturing attitude may be the best medicine and lead to a rewarding rapport where an otherwise challenging disease exists.
Prognosis
There is limited data on the prognosis of POTS patients. Recent short term follow-up studies have shown better prognosis in patients with POTS. 21 Roughly 50% of post-viral POTS patients make a meaningful recovery over about 2-5 years. Meaningful recovery may be defined as the absence of orthostatic symptoms and the ability to perform the activities of daily living with little or no restriction. Some patients experience a partial recovery and still others may demonstrate a progressive functional decline with time. As a general principle, a younger age of onset portends a better prognosis. A majority of patients tend to adopt different lifestyle modifications including increased fluid and salt intake to improve and reduce exacerbation of the POTS symptoms. In secondary POTS syndromes have a prognosis consistent with the underlying causative disorder.
Conclusion
Disruption of normal autonomic function may manifest as one of a heterogeneous group of clinical disorders collectively referred to as postural orthostatic tachycardia syndrome. Treatment is most successful when diligence has been taken to investigate the underlying disorder or POTS subtype and a comprehensive targeted treatment program is instituted with frequent follow up. Goals of care should focus on functional milestones and maintenance of function.
A 73 year old male retired civil servant with a background of spinal bulbar atrophy and hypertension presented to his General Practitioner (GP) for a routine health check. He was taking bendroflumethiazide, propranolol, atorvastatin and aspirin. His brother also has spinal bulbar atrophy.
The GP sent routine blood tests, which came back as follows: Haemoglobin 8.5 (13-17g/dL), Mean Cell Volume 84.9 (80-100fl), White Cell Count 3.4 (4-11 x10^9/L), Neutrophil Count 0.68 (2-8 x10^9/L), Platelets 19 (150-400 x10^9/L). A random blood sugar reading was 18 (3.9-7.8mmol/L). Renal function, bone profile and hepatic function tests were normal. The General Practitioner referred the patient urgently to the local Haematology unit for further assessment.
On further review the patient complained of tiredness but had had no infections or bleeding. There were no night sweats or recent foreign travel. Physical examination was unremarkable, with no lymphadenopathy or organomegaly.
A blood film showed marked anaemia with red cell anisopoikilocytosis, prominent tear drop cells and neutropenia with normal white cell morphology. There were no platelet clumps. A diagnostic investigation followed.
QUESTIONS
What are the differential diagnoses of pancytopenia and which causes are likely here given the findings on examination of the peripheral blood film?
Infections - Viral infections including cytomegalovirus, hepatitis A-E, Epstein-Barr virus, Parvovirus B19 and non A-E hepatitis viruses can cause aplastic anaemia1. The classical picture would be pancytopenia in a young patient who has recently had ‘slapped cheek syndrome’ from parvovirus B19 who has transient bone marrow aplasia. Tropical infections such as visceral leishmania may cause pancytopenia, splenomegaly and a polyclonal rise in immunoglobulins2. Overwhelming sepsis may also cause pancytopenia with a leucoerythroblastic blood film (myeloid precursors, nucleated red blood cells and tear drop red cells). HIV is also an important cause of cytopenias.
Medications - common medications may cause aplastic anaemia, such as chloramphenicol, azathioprine and sodium valproate. The history in this case did not have any recent medications introduced. The other very common cause of pancytopenia in modern practice would be in the context of chemotherapy.
Bone marrow disorders - tear drop cells are a key finding and clue in this case. They suggest an underlying bone marrow disorder and stress. In the context of a known active malignancy they are nearly indicative of bony metastases. Our patient did not have a known malignancy and there was nothing to suggest this on the history or physical examination, although in a man of this age metastatic prostate cancer should be considered. Other bone marrow disorders that would need to be considered are acute leukaemia (which was the diagnosis here), myelodysplasia and myelofibrosis. Splenomegaly would be especially significant in this case as it would be highly suggestive of myelofibrosis in combination with tear drop cells wit pancytopenia in an elderly patient3.
B12 and folate deficiency – this may cause pancytopenia, tear drop cells and leucoerythroblastic blood findings4&5. The mean corpuscular volume in this case however is normal which would somewhat argue against B12 and folate deficiency, as well as the fact that there were no hypersegmented neutrophils seen on the blood film. This cause is however very important given how it is easily reversible and treatable.
Haemophagocytosis – this is a bone marrow manifestation of severe inflammation and is a manifestation of systemic disease6. It has various causes including viruses (e.g. Epstein Barr virus), malignancy and autoimmune disease. It should be considered in patients with prolonged fever, splenomegaly and cytopenias. It is diagnosed by characteristic findings on bone marrow biopsy.
Paroxysmal nocturnal haemoglobinuria – this is a triad of pancytopenia, thrombosis and haemolysis caused by a clonal stem cell disorder with loss of membrane proteins (e.g. CD55 and CD59) that prevent complement activation7.
Genetic disease – Fanconi anaemia is a rare autosomal recessive disease with progressive pancytopenia, malignancy and developmental delay. It is caused be defects in DNA repair genes.
The key finding in this case was tear drop cells on the blood film. These are part of a leucoerythroblastic blood picture seen in bone marrow disease, malignant marrow infiltration, systemic illness and occasionally haematinic deficiency. See above for why this is unlikely to be haematinic deficiency. Although tear drop cells can occur in systemic illness such as severe infection, the history here was not in keeping with this. The diagnoses remaining therefore are malignant bone marrow infiltration or a primary bone marrow disorder (myelodysplasia, acute leukaemia or myelofibrosis). There were no features in the history pointing towards a metastatic malignancy and therefore primary bone marrow disorder is the most likely diagnosis. The diagnosis was later established as acute myeloid leukaemia on bone marrow examination.
What investigations would help to confirm or eliminate the possible diagnoses?
Blood tests including a clotting screen, liver function tests, inflammatory markers and renal function shall help to exclude other systemic disease such as disseminated intravascular coagulation, sepsis, liver disease and thrombotic thrombocytopenic purpura which may all give rise to cytopenias. Autoimmune screening may also suggest vasculitis which can cause cytopenias.
Microbiology studies including virology tests (e.g. human immunodeficiency virus, Epstein Barr virus and hepatitis viruses) may also be requested as appropriate given the clinical scenario and findings. Visceral leishmania should be tested for according to travel history and clinical likelihood. Leishmania may be identified through serology and light microscopy (for amastigotes) or polymerase chain reaction of the bone marrow aspirate. Tuberculosis could be cultured from the bone marrow is suspected.
Haematinics are a crucial test and the aim should be to try and withhold transfusion until these results are known in case they can easily be replaced thereby negating the need for blood products. Remember that if haematinics are not tested before transfusion then the blood products will confound the tests results.
Bone marrow biopsy, including aspirate and trephine are a crucial investigation for morphological examination and microbiological testing if indicated. This will distinguish the bone marrow disorders including acute leukaemia, myelofibrosis, bone marrow metastatic infiltration and myelodysplasia. Haemophagocytic syndrome may also be suggested by bone marrow examination findings.
Imaging if there is suspicion of an underlying malignancy (e.g. CT chest, abdomen and pelvis) and then further blood tests such as the prostate specific antigen. Ultrasound could also be used to check for splenomegaly where clinical examination has not been conclusive.
Medication review is vital as this may reveal the diagnosis (e.g. use of chloramphenicol)
Flow cytometry may be considered to investigate for an abnormal clone in the case of paroxysmal nocturnal haemoglobinuria and may be used on bone marrow samples to further evaluate the cells.
Unless a very clear cause for the pancytopenia is obvious (e.g. haematinic deficiency or malignant infiltration) then bone marrow examination is crucial for establishing a diagnosis. This will also prevent inappropriate treatments being initiated.
What immediate management steps and advice would be given to this patient?
General measures for pancytopenia include blood product support. Red cells and platelets can be given for symptomatic anaemia and bleeding. There is no need to transfuse platelets in the patient if there are no signs of bleeding. Alternatively he could also be treated with tranexamic acid as an alternative to avoiding risks associated with platelet transfusion. Infection should be treated urgently. Due to the neutropenia he should be advised to seek medical help if he develops a fever or sore throat. He should urgently be followed up in clinic with the results and given the contact details for the haematology department in the interim period in case he develops any problems.
The specific treatments for pancytopenia rests on the exact cause found after investigation. In this case the diagnosis was acute myeloid leukaemia arising from a background of myelodysplasia. The treatment for acute myeloid leukaemia in general, with curative intent, would consist of induction chemotherapy with DA (daunorubicin and cytosine arabinoside) followed by consolidation with further chemotherapy, the type of which (e.g. high dose cytosine arabinoside or FLAG-Ida) would depend on the risk assessment of the disease and possible consideration of an allograft bone marrow transplant after consolidation. Currently different approaches to consolidation chemotherapy, transplantation and small molecule inhibitors are being evaluated in clinical trial (e.g. AML 17 clinical trial).
The other options, in older more frail patients where high dose chemotherapy will be very toxic, are low dose palliative chemotherapy and support with transfusion.
PATIENT OUTCOME
He has been supported with blood products (platelets and packed red cells for bleeding and anaemia respectively). After discussion with him and his wife he has elected to have palliative chemotherapy with low dose cytosine arabinoside. He will be seen regularly in the haematology clinic and day unit for review. We do not suspect a link between the leukaemia and spinal bulbar atrophy.
In traumatic brain injury (TBI) the primary insult to the brain and the secondary insults as a result of systemic complications may result in a multitude of sequelae ranging from subtle neurological deficits to significant morbidity and mortality. As the brain recovers by repair and adaptation, changes become apparent and may result in physical, cognitive and psychosocial dysfunction. Rehabilitation is usually structured to recover physical ability, cognitive and social retraining with the aim of gaining independence in activities of daily living.
Case Report:
A 76 year old male patient was admitted to an intermediate neurorehabilitation unit following a traumatic brain injury(TBI) .He had fallen from a height of 11 feet resulting in intracerebral haemorrhage in the left parietal lobe and a left parietotemopral subarachnoid hemorrhage which was managed conservatively in the neurosurgical unit. He developed recurrent post traumatic seizures in the form of myoclonic jerks for which he was started on antiepileptic drugs (AEDs) sodium valproate ,clobazam and levetiracetam .During his stay in the acute neurorehabilitation unit, he was noted to be confused and wandering with a disrupted sleep wake cycle. Cognitive assessment showed global impairment across all cognitive domains suggesting that cognitive impairment was secondary to TBI with the chaotic sleeping pattern and fatigue having a significant effect on his cognition. He was then transferred to an intermediate neurorehabilitation unit four months post head injury for rehabilitation prior to discharge.
On admission he was confused, and disorientated. His neurological examination was normal except for mild expressive dysphasia. On the first night of his stay in the unit, he did not sleep at all, was restless, agitated and aggressive towards the staff. His initial agitation was attributed to the change of surroundings and general disorientation. However during his first week at the rehabilitation unit it was noted that his sleep wake cycle was completely disrupted .He would have short fragmented naps through the day and would regularly get agitated at night with threatening behaviour towards staff. On admission the Rancho Los Amigos scale† was 4(confused-agitated) and he needed specialized supervision. Despite environmental modification and optimal pharmacotherapy to improve sleep and decrease agitation, the patient still continued to have aggressive outbursts and no identifiable sleep wake pattern. It was noted by the nursing staff that occasionally when very agitated, the patient refused to have his night time medications including all AEDs .On such occasions he was reported to have slept better at night and did not have any daytime naps. All blood investigations were within normal limits except for mild hyponatremia with a normal creatinine clearance and CT head showed changes consistent with previous TBI with no new pathology .A neurology opinion was sought and with a Naranjo adverse drug reaction probability score†† of 7/10, a decision was taken to slowly decrease levetiracetam and wean it to stop, while continuing all other regular AEDs. The levetiracetam was reduced from an original dose of 750mg twice daily by 500mg every week with an aim to stop. This resulted in a considerable improvement in the patient’s agitation with a complete halt in the nighttime aggressiveness. His sleep wake cycle normalized and he started sleeping longer at night. His Ranchos Los Amigos scale improved from 4(confused-agitated) to 6(confused-appropriate). The patient could now participate more with the team of trained therapists in memory and attention exercises as well as regaining independence in activities of daily living.
Discussion:
TBI particularly in elderly aged over 64 years has a worse functional outcome as compared to non elderly.1Closed head injury in older adults produces considerable cognitive deficits in the early stages of recovery2 and there have been studies suggesting TBI to be a risk factor for developing Alzheimer’s disease.3Memory deficits, attention problems, loss of executive function and confusion are common after TBI.4This impaired cognitive function reduces the patient’s ability to recognize environmental stimuli often resulting in agitation and aggression towards perceived threats.TBI by itself may result in a variety of sleep disorders ranging from hypersomnia, narcolepsy, alteration of sleep wake cycle, insomnia to movement disorders. 5Sleep wake schedule disorders following TBI are relatively rare and may clinically present as insomnia.6Often these sleep disorders result in additional neurocognitive deficits and functional impairment, which might often be attributed to the original brain injury itself and thus be left without specific treatment.
While dealing with disrupted sleep pattern and agitation in the elderly following TBI, treatable causes such as neurological, infectious, metabolic, and medications should be ruled out. This is imperative as they disrupt rehabilitation and achievement of functional goals. Long duration of agitation post TBI has been associated with longer duration of rehabilitation stay and persisting limitations in functional independence.7After ruling out all the treatable causes the first focus is on environmental management with provision of a safe, quiet, familiar, structured environment while reducing stimulation and providing emotional support. The next step is introduction of pharmacotherapy to reduce agitation. Though a variety of pharmacological agents are available, there is no firm evidence of efficacy of any one class and often the choice of drug is decided by monitoring its effectiveness in practice and watching for side-effects.8In pharmacotherapy, the general principle followed is start low and go slow while developing clear goals to help decide when to wean and stop medications. Atypical antipsychotics are often used for the agitation while benzodiazepines and non benzodiazepine hypnotics such as zopiclone are recommended for treatment of insomnia.9 However atypical antipsychotics carry a FDA black box warning being associated with increased risk of stroke and death among elderly.
But what does one do when all optimal non pharmacologic and pharmacologic measures fail? That brings us back to the drawing board which in this case led the team to rethink Levetiracetam, a novel new antiepileptic that has been used as monotherapy for partial seizures and adjunctive therapy for generalized tonic clonic and myoclonic seizures. Levetiracetam treated patients have been reported to have psychiatric adverse effects10 including agitation, hostility, anxiety, apathy, emotional lability, depersonalization, and depressionwith few case reports of frank psychosis 11.While in healthy volunteers levetiracetam is noted to consolidate sleep12,in patients with complex partial seizures, levetiracetam has been noted to cause drowsiness decreasing day time motor activity and increasing naps without any major effects on total sleep time and sleep efficiency during night.13There has been an isolated report of psychic disturbances following administration of levetiracetam and valproate in a patient with epilepsy which resolved following withdrawal of valproate. 14However in practice it is used for recurrent post TBI seizures as it is a potent AED with a relatively mild adverse effect profile and no clinically significant interactions with commonly prescribed AEDs.15
Any adverse drug reaction (ADR) should be evaluated while keeping the patients clinical state in mind. This was, indeed, difficult in our case. With a history of TBI and cognitive decline, it became difficult to ascertain whether the neurocognitive issues were purely due to the nature of TBI or due to an ADR. Assigning causality to a single agent is difficult and fraught with errors. Using the Naranjo algorithm 16, with a score of 7/10(probable ADR) and a notable response on withdrawal of the offending drug as in this case helps establish possible causality.
This is a rare instance where sleep wake cycle disorder and agitation resolved following withdrawal of Levetiracetam in an elderly patient with TBI. This in turn led to the patient having a stable mood so that therapists could communicate and interact with him in order to improve basic cognitive functions such as attention, memory, thinking and executive control. This case illustrates the constant need to systematically and frequently reassess patients as they recover from TBI.
†Appendix: Ranchos Los Amigos Levels of cognitive functioning.
Fibromyalgia (FM) is a challenging set of chronic, overlapping and debilitating syndromes with widespread pain, abnormal pain processing, sleep disturbance, fatigue and psychological distress.1 The American College of Rheumatology (ACR) 1990 diagnostic guidelines were based primarily on tender point examination findings at 11 of 18 potential tender points;2 however, lack of consistent application of these guidelines in clinical settings led the ACR in 2010 to develop new diagnostic criteria based on a Widespread Pain Index (WPI) and symptom severity (SS) scale with no requirement of a tender point examination. Symptoms must have been present for at least three months with the absence of any other disorder that would otherwise explain the pain and other signs and symptoms.3
Type of pain and other symptoms vary widely in FM, complicating diagnosis and treatment. A cross-sectional survey of 3,035 patients in Germany utilized cluster analysis to evaluate daily records of symptoms noted by patients on handheld computers. Five subgroups were described: four with pain evoked by thermal stimuli, spontaneous burning pain, pressure pain, and pressure pain combined with spontaneous pain; the fifth subgroup had moderate sensory disturbances, but greater sleep disturbances and the highest depression scores.4
Estimates of the prevalence of FM have varied based on case definitions and survey methods. Using 1990 ACR guidelines, it was estimated to affect between 0.1 to 3.3% of populations in western countries and 2.0% in the United States. Greater prevalence occurs among females, with estimates ranging from 1.0 to 4.9%.1, 5 Reasons for the gender difference have not been determined.6-9
Fibromyalgia Risk Factors
Identification of risk factors for FM has been complicated by the array of seemingly unrelated signs and symptoms. The United States Centers for Disease Control (CDC) notes loose association with genetic predisposition,10 bacterial and viral infections, toxins, allergies, autoimmunity, obesity and both physical and emotional trauma.1, 11
Chronic fatigue syndrome and infection
Although chronic fatigue syndrome (CFS) has been defined as a separate syndrome, up to 70% of patients with FM are also diagnosed with CFS and 35-70% of patients with CFS have also been diagnosed with FM.12 Thus studies of patients with CFS may have clinical relevance to FM. Several case controlled studies of CFS and one of CFS/FM have been associated with chronic bacterial infections due to Chlamydia (Chlamydophila p.), Mycoplasma, Brucella, and Borrelia.12-18 The most prevalent chronic infection found has been that of the various Mycoplasmaspecies.15-23
Mycoplasmas are commonly found in the mucosa of the oral cavity, intestinal and urogenital tracts, but risk of systemic illness occurs with invasion into the blood vascular system and subsequent colonization of organs and other tissues.15-23Mycoplasmal infections have been identified in 52 – 70% of CFS patients compared with 5 to 10% of healthy subjects in North America15-17, 19-22 and Europe (Belgium)23. For example, the odds ratio (OR) of finding Mycoplasma species in CFS was 13.8 (95% CL 5.8-32.9, p< 0.001) in North America.17A review by Endresen12 concluded that mycoplasmal blood infection could be detected in about 50% of patients with CFS and/or FM. A CDC case-control study attempted to replicate these findings based on the hypothesis that intracellular bacteria would leave some evidence of cellular debris in cell-free plasma samples. Results were that the healthy subjects actually had evidence of more bacteria although the difference was not significant. The authors noted the complexity and limitations of this type of analysis and also postulated that since the CFS patients were years past the onset of illness, they might have previously cleared the triggering agent.24 However, most studies found Mycoplasma DNA in intracellular but not extracellular compartments in CFS patients, and this could explain the discrepancy.15-23 Other studies have found that 10.8% of CFS patients were positive for Brucella species (OR=8.2, 95% CL 1-66, p<0.01)16 and 8% werepositive for Chlamydia pn. (OR= 8.6; 95% CL 1-71.1,p< 0.01)17.
The presence of multiple co-infections may be an especially critical factor associated with either initiation or progression of CFS. Multiple infections have been found in about one-half of Mycoplasma-positive CFS patients (OR = 18.0, 95% CL 8.5-37.9, p< 0.001), compared with single infections in the few control subjects with any evidence of infection.17 A North American study identified chronic infections in 142 of 200 patients (71%) with 22% of all patients having multiple mycoplasmal infections while just 12 of the 100 control subjects (12%) had infections (p<0.01) and none had multiple infections.15 Similarly, a European study reported chronic mycoplasmal infections in 68.6% of CFS and 5.6% of controls. Multiple infections were found in 17.2% of the CFS patients compared with none in the controls (p<0.001).23 Multiple co-infections were also associated with significantly increased severity of symptoms (p<0.01).15, 23
Viral infections associated with CFS have included Epstein Barr virus, human herpes virus-6, cytomegalovirus, enteroviruses and several other viruses.15, 25, 26
Despite indications of single or multiple bacterial and/or viral infections in most patients with CFS, antibiotic or antiviral treatments have yielded inconsistent results.27 Slow growing intracellular bacteria are relatively insensitive to most antibiotics and have inactive phases when they would be completely insensitive to any antibiotics.2823 Some treatments may actually have resolved the infections, but not the immune pathways that may remain in an activated state capable of producing symptoms.
Fibromyalgia and infection
Bacterial infections associated with FM as a separate syndrome have included small intestinal bacterial overgrowth (SIBO)29, 30 and helicobacter pylori (HP)31. Utilizing the lactulose hydrogen breath test (LHBT), investigators found SIBO in 100% of 42 patients with FM. They noted that 30-75% of patients with FM have also been found to have irritable bowel syndrome (IBS).29, 30 A confounding factor is that medications prescribed for FM often have gastrointestinal side effects.29 HP diagnosed by positive immunoglobulin gamma (IgG) serum antibody was significantly higher in women with FM (44/65 or 67.7%) compared with controls (18/41 or 43.9%) (p=0.025) in Turkey31.
Viral infections associated with FM have included hepatitis C, in which two studies found an association,32-34 and two studies found no association.35, 36 Associations with FM have also been found with hepatitis B, 37 human immunodeficiency virus (HIV)38, 39 and human T cell lymphotropic virus type I (HTLV-1).40
Fibromyalgia and non-infectious associations
Non-infectious triggers associated with FM have included toxins, allergens, and physical or emotional trauma. These triggers may not have been strictly “non-infectious” as allergens and toxins may also be produced by infections, and physical or emotional trauma may lead to the reactivation of previously controlled infections. Respondents to an internet survey of people with FM (n=2,596) also identified triggers as chronic stress (41.9%), emotional trauma (31.3%), acute illness (26.7%) and accidents (motor vehicle 16.1%, non-motor vehicle 17.1%).41 Physical trauma associated with FM has included cervical spine injuries as well as motor vehicle and other accidents.42-44
Fibromyalgia and autoimmunity
Three studies have found thyroid autoantibodies to be in greater percentages in subjects with FM compared with controls, in spite of normal thyroid hormone levels. One study reported autoantibodies in 41% of FM patients versus 15% of controls.45 The second study reported 16% in FM versus 7.3% in controls, p<0.01.46 The third study reported 34.4% in FM versus 18.8% in controls (p=0.025)47 and OR =3.87, 95% CL 1.54-10.13.48 This could also have been the result of thyroiditis, because infections like Mycoplasma are often found in thyroiditis patients.15
Autoantibodies to serotonin were identified in 74% of 50 patients with FM compared with 6% of 32 healthy (blood donor) controls. Notably, serotonin levels were normal in 90% of the FM patients indicating serotonin receptor involvement.49
Fibromyalgia and Metabolic Syndrome
Metabolic Syndrome consisting of abdominal obesity, high triglycerides, high blood pressure, elevated fasting glucose and decreased high-density lipids, was associated with FM in a U.S. study in which cases were 5.6 times as likely to have Metabolic Syndrome as controls (C2MH = 3.84, p = .047, 95% CL 1.25 – 24.74).50
Fibromyalgia and emotional trauma
Although emotional trauma has been acknowledged as a contributing factor, most studies of CFS/FM have used recognized tests such as Beck’s Depression Index, Beck’s Anxiety Index and Minnesota Multi Personality Index (MMPI) to exclude potential subjects with actual psychiatric illnesses.51, 52
Psychological and physiological subsets of fibromyalgia
A Wisconsin cross sectional survey of 107 women with confirmed diagnoses of FM used validated psychological and physiological measures followed by cluster analysis. Four distinct subsets were identified: (I) history of childhood maltreatment and hypocortisolism with the most pain and disability; (II) “physiological dysregulation” described as “distinctive on nearly every biological index measured” with high levels of pain, fatigue and disability; (III) normal biomarkers with intermediate pain severity and higher global functioning; and (IV) psychological well-being with less disability and pain.53
The “physiological dysregulation” of FM subset II consisted of the highest antinuclear antibody (ANA) titers (t=4.06, p=0.001), highest total cholesterol levels (t=3.96, p<0.001), larger body mass index (BMI) values t=2.21, p<0.04), lowest Natural Killer (NK) cell numbers (t=3.95, p<0.001), lowest growth hormone (t=3.20, p<0.002), and lowest testosterone levels (t=3.80, p<0.001). Trends were also indicated toward the highest erythrocyte sedimentation rate (ESR) (t=2.02, p=0.056), lowest creatinine clearance (t=1.85, p=0.067) and lowest cortisol (t=2.78, p<0.007).53
Proposed Model of Fibromyalgia
The authors’ proposed model of FM develops a rationale for the “physiological dysregulation” indicated in subset II of the Wisconsin study. In this model, various triggers are followed by prolonged immune activation with subsequent multiple hormonal repression, disrupted collagen physiology and neuropathic pain.
Activation of immune response pathways
Innate immune responses begin with anatomical barriers, such as the epithelium and mucosal layers of the gastrointestinal, urogenital and respiratory tracts, and physiological barriers, such as the low pH of stomach acid and hydrolytic enzymes in bodily secretions. 54 Breeching of these barriers activates cell-mediated immunity launched by leucocytes with pattern recognition receptors: neutrophils, macrophages and dendritic cells (DCs).54 Insufficient or damaged anatomical or physiological barriers would necessarily keep this cell mediated level of innate defense in a constant state of alert and activity.
In contrast to the innate immune response, adaptive immunity has highly specific recognition and response activities resulting in lasting changes produced by leukocytes known as lymphocytes. B lymphocytes (B cells) secrete plasma cells producing antibodies to specific pathogens. T lymphocytes (T cells), the other major cells of adaptive immunity, can be either cytotoxic (Tc) or helper cells (Th). Tc cells produce progeny that are toxic to non-self peptides and Th lymphocytes secrete small proteins (cytokines) that mediate signaling between leukocytes and other cell types. All types of lymphocytes retain memory so that subsequent invasions provoke faster and more rapid differentiation into effector cells. 54, 55 SomeTh cells respond to intracellular pathogens (Th1) and some to extracellular pathogens (Th2). A third type (Th17) appears to respond to certain bacterial and fungal infections, tumor cells and are also involved in autoimmune diseases.56
In the presence of environmental stressors, cells may release stress proteins to alert the organism to potentially damaging conditions. These proteins can bind to peptides and other proteins to facilitate surveillance of both the intracellular and extracellular protein environment. One form of stress proteins, heat shock proteins (HSP), can mimic the effects of inflammation and can be microbicidal.52, 57
One of the earliest responses to intracellular viral or bacterial infections involves production of three types of interferon (IFNa, IFNb and IFNg). Any of these can initiate a series of metabolic events in uninfected host cells that produce an antiviral or anti-bacterial state.58, 59 When IFN-γ targets genes in uninfected cells, the targeted genes become microbicidal by encoding enzymes generating oxygen (O2) and nitric oxide (NO) radicals.58 Activation of O2 or NO radicals triggers another cascade involving IL-6, IL-1b, the cytokine Tumor Necrosis Factor-a (TNF-a) and the transcription nuclear factor kB (IKKb-NF-kB). NF-kB can be activated by a variety of inflammatory stimuli, such as cytokines, growth factors, hormones, oncogenes, viruses and their products, bacteria and fungi and their products, eukaryotic parasites, oxidative and chemical stresses, therapeutic and recreational drugs, additional chemical agents, natural products, and physical and psychological stresses.60 Activation of NF-kB releases its subunits; the p50 subunit has been associated with autoimmunity and the RelA/p65 unit with transcriptional activity involving cell adhesion molecules, cytokines, hematopoietic growth factors, acute phase proteins, transcription factors and viral genes.61 The authors propose that chronic infection or other stress would be a sustaining trigger of an immune cascade that includes NF-kB and resultant cell signaling processes that drive many of the symptoms of fibromyalgia.
The cytokine interleukin-6 (IL-6) can either activate or repress NF-kB through a switching mechanism involving IL-1ra and Interleukin 1b(IL-1b). IL-6 first activates Interleukin 1b (IL-1b), which then activates TNF-a, leading to the subsequent activation of NF-kB. 62, 63 Specifically, the release of the RelA/p65 subunit of activated NF-kB switches on an inhibitory signaling protein gene (Smad 7) that blocks phosphorylation of Transforming Growth Factor Beta (TGF-b) resulting in the repression of multiple genes. Alternatively, IL-6 activates IL-1ra, which allows TGF-b to phosphorylate and induce the expression of activating signaling protein genes Smad2 and Smad3, resulting in the full expression of multiple genes.61
NF-kB plays a key role in the development and maintenance of intra- (Th1) and inter- (Th2) cellular immunity through the regulation of developing B and T lymphocytes. The p50 dimer of NF-kB has been shown to block B Cell Receptor (BCR) editing in macrophages, resulting in loss of recognition and tolerance of host cells (autoimmunity). T cells that are strongly auto-reactive are normally eliminated in the thymus, but weakly reactive ones are allowed to survive to be subsequently regulated by regulatory T-cells and macrophages. Acquired defects in peripheral T-regulatory cells may mean failure to recognize and eliminate weakly reactive ones.54, 64 The IL-17 cytokine associated with autoimmunity can activate NF-kB through a pathway that does not require TNF-a.56 NF-kB activity can also be activated or repressed by the conversion of adenosine triphosphate (ATP) to cyclic adenosine monophosphate (c AMP) in the early phases (3 days) of nerve injury through its main effector enzyme, protein kinase A (PKA).65, 66 PKA decreases during later stages as the enzyme protein kinase C (PKC) increases. PKC then plays important roles in several cell type specific signal transduction cascades.67 An isoform of PKC within primary afferent nociceptive nerve fibers signals through IL-1b and prostaglandins E2 (PGE2) as demonstrated in animal studies.68 This process has been called “hyperalgesic priming,” and it has been described as responsible for the switch from acute to long-lasting hypersensitivity to inflammatory cytokines.69
Figure 1 depicts key immune pathways leading to expression or repression of multiple genes proposed to be important in FM and neuropathic pain.
Fibromyalgia and immune - hormonal interactions
Reciprocity exists between the immune system and the hypothalamic-pituitary-adrenal (HPA) axis through its production of glucocorticoid signal transduction cascades. 63, 70, 71. Hormones such as cortisol (hydrocortisone) produced by the adrenal cortex, affect metabolism of glucose, fat and protein.72 The glucocorticoid receptor (GR), a member of the steroid/thyroid/retinoid super family of nuclear receptors is expressed in “virtually all cells”. When the GR in the cytoplasm binds a glucocorticoid, it migrates to the nucleus where it modulates gene transcription resulting in either expression or repression of TNF-a, IL-1bβ and the NF-kB p65/RelA subunit. However, the RelA/p65 protein can also repress the Glucocorticoid Receptor. 63, 70, 71, 73
Growth hormone (GH), an activator of NF-kB,74 is usually secreted by the anterior pituitary, but changes found in FM may be hypothalamic in origin. GH is needed for normal childhood growth and adult recovery from physical stresses.75 Although low levels of GH were found in subset II of the Wisconsin study, 53 functional deficiency may be expressed as low insulin-like growth factor 1 (IGF-1) combined with elevated GH, suggesting GH resistance.76, 77 Defective GH response to exercise has been associated with increased pain and elevated levels of IL-1b, IL-6, and IL-8.77, 78
The hormones serotonin and norepinephrine modulate the movement of pain signals within the brain. Serotonin has been found to suppress inflammatory cytokine generation by human monocytes through inhibition of the NF-kB cytokine pathway in vitro;79 however, NF-kB promotion of antibodies can repress serotonin.49 Selective serotonin and norepinephrine reuptake inhibitors (SSNRIs), such as duloxetine and milnacipran, are key treatment options for fibromyalgia and have been approved of by the U.S. Food and Drug Administration (FDA).80, 81 Although serotonin has been best measured in cerebral spinal fluid (CSF), recently improved methods of collection were utilized (using rats and in 18 women) that yielded a high degree of correlation (r=0.97) between CSF and plasma, platelet, and urine measurements.82
NF-kB activation has also been documented to interfere with thyroid hormone action through impairment of Triiodothyronine (T3) gene expression in hepatic cells. 83 However, T3 administration has induced oxidative stress and activated NF-kB in rats.84
Metabolic Syndrome, a confounding factor in Fibromyalgia
Leptin and insulin hormones interact to regulate appetite and energy metabolism. Leptin, produced by adipose cells, circulates in the blood eventually crossing the blood-brain barrier to bond with a network of receptors within the hypothalamus. Insulin, produced by beta cells in the pancreas, similarly crosses the blood brain barrier to interact with its own network of hypothalamic receptors. Leptin and its receptors share structural and functional similarities to long-chain helical cytokines, such as IL-6, and it has been suggested that leptin be classified as a cytokine.85-89
Metabolic syndrome can be a confounding factor in FM due to peripheral accumulation of fatty acids, acylglycerols and lipid intermediates in liver, bone, skeletal muscle and endothelial cells. This promotes oxidative endoplasmic reticulum (ER) stress and the activation of inflammatory pathways involving PKC and hypothalamic NF-kB, leading to central insulin and leptin repression.85-87, 89-91Hyperinsulinemia further stimulates adipose cells to secrete and attract cytokines such as TNFa and IL-6 that trigger NF-kB in a positive feedback loop, which can be complicated by chronic over nutrition that increases the generation of reactive oxygen intermediates and monocytechemoattractant protein-1 (MCP-1).87, 89 When exposed to a chronic high fat diet, hypothalamic NF-kB was activated two fold in normal mice and six times in mice with the obese (OB) gene.89
Fibromyalgia and indicators of immune-hormonal activity
Although most components of either innate or adaptive cell mediated immune responses exist for only fractions of seconds, some of their effects and products can be detected long after in the skin, muscle, blood, saliva or sweat92, 93.
One component, nitric oxide (NO), can suppress bacteria; however, endothelial damage causes dysfunction with impaired release of NO and loss of its protective properties.86 The enzyme transaldolase acts as a counterbalance by limiting NO damage to normal cells. Thus, high levels of transaldolase indicate elevated reactive oxygen species, reactive nitrogen species (ROS/RNS) and cellular stress. The “exclusive and significant over-expression of transaldolase” in the saliva samples of 22 women with FM compared with 26 healthy controls (77.3% sensitivity and 84.6% specificity, p<0.0001; 3 times greater than controls; p=0.02) was “the most relevant observation”; although there was no correlation between transaldolase expression and the severity of FM symptoms.92
High levels of NO have been associated with high levels of insulin, and insulin itself is a vasodilator that, in turn, can stimulate NO production. Beta cells of the pancreas are quite susceptible to ROS/NOS damage .86When free radical damage of beta cells reaches critical mass, insulin production plummets with an associated decline in NO levels. Thus, patients with FM who have high NO levels would likely be suffering from associated metabolic syndrome, and patients with low NO levels would likely be suffering from Type II diabetes.85, 88
Figure 2 illustrates the relationship of NF-kB to various hormone systems.
Fibromyalgia and immune-hormonal influences on connective tissue
Inflammation of muscles, tendons, and/or fascia is generally followed by proliferative and remodeling phases of healing initiated by fibroblasts which lay down an extracellular matrix (ECM) composed of collagen and elastin fibers. “Fibroblasts respond to mechanical strain by altering shape and alignment, undergoing hyperplasia and secreting inflammatory cytokines including IL-6.” The extra cellular matrix is initially laid down in a disorganized pattern that is subsequently matured and aligned. Chronic and excessive mechanical tension from postural imbalance, hormonal disruption or other factors may interfere with collagen maturation. 94 Remodeling of the extracellular matrix and collagen deposition around terminal nerve fibers may be compressive and contribute to neuropathic pain.95
Oxidative stress in muscles accelerates the generation of advanced glucose (glycation) end products (AGEs). AGE-mediated cross-linked proteins have decreased solubility and are highly resistant to proteolytic digestion. Interaction of AGEs with their receptors leads to activation of NF-kB resulting in an increased expression of cytokines, chemokines, growth factors, and adhesion molecules.9697
Two AGE products have been reported at significantly elevated levels in the serum of patients with FM: N-carboxymethyllysine (CML) (2386.56 ± 73.48 pmol/mL; CL 61.36-2611.76 versus controls 2121.97 ± 459.41 pmol/mL; CL 2020.39-2223.560; p<0.05)96 and pentosidine (mean 190 ± 120 SD and median 164 versus controls mean 128 ± 37 SD and median 124; p<0.05)97 Comparison of muscle biopsies showed “clear differences in the intensity and distribution of the immunohistochemical staining”. CML was seen primarily in the interstitial tissue between the muscle fibers where collagens were localized and in the endothelium of small vessels of patients. Activated NF-kB was seen in cells of the interstitial tissue especially around the vessels of patients, but almost no activated NF-kB was seen in the control biopsies. AGE activation of NF-kB has been shown to be significantly more prolonged than the activation of NF-kB by cytokines.9697
Fibromyalgia, the nervous system and pain
Sensory transmission in humans occurs through three primary afferentnerve fiber types: heavily myelinated mechanical afferent pathways (A Beta fibers) that transmit non-noxious tactile sensations, small-diameter myelinated fibers (A Delta fibers) that transmit sharp pain, and small diameter unmyelinated fibers (C fibers) that transmit dull aching pain. The heavily myelinated non-pain Aβ fiber type has been shown to sprout axons that terminate on pain lamina in the posterior horn of the spinal cord resulting in the conversion of mechanical stimuli to pain. Within the brain, sensitization of the N-methyl D-aspartate (NMDA) receptors can amplify pain signals between the thalamus and the sensory cortex.67, 98
Chronic damage or excitation of nociceptive afferent fibers from compressive collagen deposition may develop into spontaneous (ectopic) firing oscillating at frequencies sufficient to initiate cross (ephaptic) excitation of sympathetic and sensory fibers (myelinated A-delta and non-myelinated C fibers) within the dorsal root ganglia (DRG) of the central nervous system.98 Normally, the DRG has little sympathetic innervation, but trauma can trigger sympathetic sprouting that forms basket-like structures within the DRG. Neurotrophins, in particular nerve growth factor (NGF), play an important role in sympathetic fiber sprouting of sensory ganglia in murine models. DRG can be reservoirs for latent viral infections such as Herpes Zoster, HIV and enteroviruses. In addition, the Borrelia species has been identified in a non-human primate model of Lyme disease. NGF also facilitates expression of Substance P (SP), a peptide neurotransmitter involved in the induction of the IL-6 - NF-kB pathway 60, 99, 100 and in the transmission of neuropathic pain.101, 102 SP has been found to be elevated in the cerebrospinal fluid of patients with FM in comparison to normal values,103 and control subjects.104
Summary and Conclusions
Chronic unresolved infection, trauma, and/or emotional stresses that trigger immune pathways with subsequent chronic hormonal and nervous system responses is proposed to perpetuate chronic neuropathic pain. Figure 3 provides a summary model of immune-hormonal contributions to neuropathic pain in fibromyalgia.
The ACR criteria and severity scales have defined fibromyalgia and The Wisconsin study has identified psychological and physiological subsets that are critical steps in its characterization. This type of testing could be further strengthened through the use of specific biomarkers. Potential markers of FM status include the RelA/p65 and p50 subunits of NF-kB, which are currently the focus of several clinical trials of other chronic painful conditions. Additional potential markers include: IL-6, IL-1b, TNF-a, PKC, transaldolase, CML, pentosidine and NGF. Substance P has been previously identified as a marker of pain, but is problematic as a marker for FM, since it has only been measured in the CSF. The search for markers that are truly specific to FM may continue to be a difficult task due to their overlap with other metabolic conditions, such as CFS, metabolic syndrome, type II diabetes, and IBS. Nonetheless, these markers remain important as they can indicate oxidative stress, cytokine activation, hormonal dysregulation and neuropathic pain. These potential FM markers need to be evaluated in clinical trials where they can be measured over time and correlated with patient symptoms.
Currently, family and general medical practice physicians are uniquely positioned to establish the FM diagnosis, determine subsets of FM patients, investigate potential triggers of chronic immune activation, advise patients, prescribe medications and refer patients to appropriate specialists or pain centers. Establishment of the FM diagnosis requires use of the ACR Widespread Pain Index (WPI) and symptom severity (SS) scale, but no longer requires the tender point examination. 3
Determination of FM subsets can be accomplished using the approach used in the Wisconsin cross sectional survey.53 Investigation of potential triggers of chronic immune activation needs to include sources of underlying infection, unresolved physical or emotional trauma, toxins and food sensitivities. These investigations may be accomplished through careful interviewing and well-designed questionnaires. Advising the patient should acknowledge the reality of their pain and other symptoms and provide rational approaches to resolution of those symptoms. Prescribing of medications needs to be sensitive to current and previous patient experience with medications, in addition to following current guidelines for stabilizing FM symptoms. Referral to appropriate specialists and centers would include those with expertise in physical medicine, psychology and nutrition. Physical medicine can address pain and functional deficits; psychology can address underlying emotional issues and trauma; and nutrition can focus on resolution of chronic inflammation, oxidative stress, and intestinal dysbiosis.
Where do we go from here for additional FM treatment options? Immune modulators have been used successfully in other painful conditions, such as rheumatoid arthritis. Immune modulators acting on the IL-6 - NF-kB cascade have considerable potential for FM, but only after ruling out or successfully treating any underlying infections. Numerous pharmaceutical blockers of NF-kB exist, but most are associated with serious side effects. Natural products may provide additional options as some are able to mediate pathways leading to NF-kB without the same side effects.105Medications that elevate individual hormone levels have been included in accepted treatment protocols in the case of serotonin and norepinephrine. However, elevations of other hormones, such as cortisol and thyroid hormones, are under investigation and remain controversial. Elevation of individual hormones may be problematic because of the number of different hormones influenced by the IL-6 - NF-kB pathway.
There are approximately over 1.6 billion overweight people with a body mass index (BMI) greater than 25 kg/mAnnually, around 2.8 million deaths are attributed to overweight and obesity worldwide(1). Many overweight individuals underestimate their weight and despite acknowledging their overweightness, many are not motivated to losing weight(2).Accurate measurement is important as it identifies patients with diagnoses which subsequently impact on their management. Self-reported weight is often used as a means of surveillance but has been shown to bias towards under reporting of body weight and BMI as well as over reporting on height(3). Several estimation techniques has been devised to quantify anthropomorphic measurements when actual measurement cannot take place(4),(5),(6), however, these methods are associated with significant errors for hospitalised patients(7). There is no published study that questions the validity of visual estimation of obesity in daily clinical setting despite its relevance to the daily practice. We aim to investigate the accuracy of visual estimation compared to actual clinical measurements in the diagnosis of overweight and obesity.
Methods:
This is a case control study. Patients for this study were attending the endocrinology, cardiology and chest pain out-patient clinic in Cork University Hospital, Cork, Ireland. The questionnaire session was carried out at every endocrinology, cardiology and chest pain clinic for 5 consecutive weeks. A total of 100 patients were recruited allowing for a 10% margin of error at 95% confidence level in a sample population of 150 000. Ten doctors of varying grades were chosen randomly to visually score the subjects. Exclusion criteria included patients who were pregnant and who are wheelchair bound. Consent was obtained from patients prior to filling questionnaires. Ethical approval was received from the Clinical Research Ethics Committee of the Cork Teaching Hospitals.
In the waiting room, patients were asked to self- report their weight, height and waist circumference to the best of their estimate. Demographics and cardiovascular risk were obtained from medical charts and presented in Table 1. The questionnaires have a section that specifically tests patients’ awareness of abdominal obesity and patients were asked to choose between obesity and abdominal obesity, relying on their own knowledge of markers of cardiovascular risks. Clinical measurements were taken in the nurses’ assessment room. Weight was measured by using portable SECA scales (Seca 755 Mechanical Column Scale) and was measured to the nearest 0.1kilogram. All patients were measured on the same weighing scale to minimize instrumental bias. Patients were asked to remove their heavy outer garments and shoes and empty their pockets and to stand in the centre of the platform, so that weight is distributed evenly to both feet.
Height was measured by using a height rule attached to a fixed measuring rod (Seca 220 Telescopic Measuring Rod). Patients were asked to remove their shoes and are asked to stand with their back to the height rule. It was ensured that the back of the head, back, buttocks, calves and heels are touching the wall. Patients were asked to remain upright with their feet together. The top of the external auditory meatus is leveled with the inferior margin of the bony orbit. The patients were asked to look straight. Height is recorded to the resolution of the height rule (i.e. nearest millimeter).
Waist circumferences were measured using a myotape. Patients were asked to remove their outer garments and stand with their feet close together. The tape is placed horizontally at a level midway between the lower rib margin and iliac crest around the body. They were then asked to breathe normally and the reading of the measurement was taken at the end of gentle exhaling. This prevents patients from holding their breath. The measuring tape is held firmly, ensuring its horizontal position and loose enough that it allows placement of one finger between the tape and the subject's body. A single operator who has been trained to measure waist circumference as per the WHO guidelines is used repeatedly in order to reduce measurement bias(8).
The doctors were asked to visually estimate the patients' weight, height, waist circumference and BMI. The estimation is recorded on a separate sheet. All doctors were blinded to the actual clinical measurements. The questionnaires were then collected at the end of the clinic and matched to individual patients. Data entry was performed in Microsoft Excel and exported for statistical analysis on SPSS version 16.
Findings
The study enrolled 100 patients. Demographic and cardiovascular risk details are shown in Table 1. Among these, 42 were obese, 35 were overweight and 23 patients had a normal BMI. The sample has a mean BMI of 29.9kg/m2 (95% CI 28.7-31.1) with a mean waist circumference (WC) of 103.2cm (95% CI 100.7-107.2). The average male waist circumference is 105.8 cm while the average female waist circumference is 101.6cm. The mean measured weight was 84.6kg (95% CI 81.0-88.2) and the mean height measurement was 1.68m (95% CI 1.66-1.70).
Table 1: Cardiovascular risk factors
Sex
Male(n=55)
Female(n=45)
Mean age
53.6(19-84)
56.7(23-84)
Diabetes
17
14
Hypertension
16
20
Hypercholesterolaemia
24
19
Active smoker
10
5
Ex- smoker (>10years)
8
3
Previous stroke or heart attack
6
6
Previous PCI
6
3
Patient’s perception and doctor’s estimation of anthropomorphic measurements were compared to actual measurements and is displayed in Table 2.
Table 2. Deviation from actual measurement values in both groups
Patient’s Estimation
Mean estimated
Mean deviation (estimated – actual measurements)
95% Confidence interval of Mean Deviation
Weight
81.16
-3.71
-5.10 to -2.32
Height
1.6782
0.0039
-0.0112 to 0.0033
Waist
90.85
-13.09
-15.48 to -10.70
BMI
28.68
-1.24
-1.87 to -0.61
Doctor’s visual estimation
Weight
80.85
-3.78
-5.54 to -2.02
Height
1.6710
-0.0113
-0.224 to 0.002
Waist
92.10
-11.84
-13.87 to -9.81
BMI
29.08
-8.47
-1.54 to -0.15
In terms of patients own estimation of height, weight and waist circumference, 49% of patients under estimated their weight by up to 1.5kg, 35% reported accurately to 1.5 kg and 16% over reported weight. 67% of patients estimated height accurately, 18% of patients under-estimated, and 15% over-estimated. When asked to estimate their waist circumference, 68% of patients under estimated by up to 5cm, 30% over estimated and 2 patients estimated accurately to 5cm (Figure 1). We found that 70% of patients regarded obesity as the higher threat to health compared to abdominal obesity. There were no differences in patient’s self reported weight and doctor’s weight estimation (p= 0.236).
Figure 1. Graphical representation of patients estimated weight, height and waist circumference
We then analysed the doctor’s estimation of height, weight, waist circumference and BMI. For the purpose of interpreting the data on BMI, the estimates that is recorded by doctors that matches the patient’s real BMI by clinical measurement is considered accurate. Therefore, for patients who have a normal BMI, 69.5% were correctly estimated as normal and the rest (30.5%) were estimated as overweight. For those patients who are obese, 81% were estimated as obese and by the doctors as a group and the rest (19%) is estimated to be overweight. In patients who are overweight, 63% were correctly estimated as being overweight by doctors, 9% were estimated as being obese and the rest (28%) were mistakenly estimated as having a normal BMI. Accurate BMI estimation by doctors was achieved in 72% patients (Figure 2).
Figure 2. Doctors estimation of BMI compared to actual clinical measurement
Doctors were noted to underestimate the patients’ weight in 53 patients, over estimated in 26, while being accurate in their estimation in 21 patients. Estimation of waist circumference to the nearest 5 cm shows marked under estimation of waist circumference in 71% of patients, over reporting in 3% of patients and 26% accurate estimation. The majority of underestimation of waist circumference happens in the region of 10 to 15cm. For patients who are obese, doctors were able to estimate waist circumference correctly in 58% of obese individuals.
Discussion:
This is the first study demonstrating the relationship of visual estimation of a cardiovascular risk factor and comparing to actual clinical measurements. As obesity and abdominal obesity becomes an increasingly common phenomenon, our perception of the 'normal' body habitus may be distorted(9).
It is observed that in the bigger hospitals out-patient departments, physicians and nurses are commonly affected by clinical workload and tend to spend a limited amount of time with patients in order to achieve a quicker turnaround time. Cleator et al looked at whether clinically significant obesity is well detected in three different outpatient department and whether they are managed appropriately once diagnosed(10). In all the outpatient departments involving the specialties of rheumatology, cardiology and orthopedics, the actual cases of clinical obesity is higher than what is being diagnosed and the management of obesity was heterogeneous and minimal in terms of intervention. With the ever increasing obese patients attending hospitals, it is understandable that healthcare providers such as physicians, nurses, dietician and physiotherapist resort to relying on visual estimation.
In terms of patient’s own estimation of height, weight and waist circumference, we gained that patients were reasonably good at estimating their own height but tend to under estimate weight. This is probably due to the fact that these patients have not had a recent measurement of weight and their weight estimation is based on previous historical measurement from months to years back, which in the majority of people, is less than their current weight. This also explains why their height estimation is more accurate, as adult heights do not undergo significant changes and are relatively constant.
When attempting to obtain patient’s own estimation of waist circumference, we found that most patients are not at all aware of the method used to measure waist circumference. Some patients even mistaken waist circumference as being their trousers’ waist size. In those who were able to give estimation, a large proportion would under estimate.
The majority of patients think that general obesity is more predictive of cardiovascular outcome compared to abdominal obesity. This lack of awareness is reflective on clinician’s effort in addressing abdominal obesity as an important cardiovascular risk factor to patients during consultations. The lack of proper awareness campaign by healthcare providers along with the evolving markers of cardiovascular risk may further confuse the general public.
Recently, waist circumference, waist to hip ratio along with many serum biomarkers have been noted to correlate to adverse outcomes in obese individuals, independent of BMI. Waist circumference measurement is a relatively new tool compared to the measurement of BMI. This would explain the discrepancy between doctors’ estimation of BMI and waist circumference. Visual estimation is further compromise as many patients would be covered in items of clothing during consultations. In order to obtain a better estimation of waist circumference, the individual have to be observed from many angles, a task that may be impossible in a busy clinic.
Although BMI is a convenient method to quantify obesity, recent studies have shown that waist circumference is a stronger predictor of cardiovascular outcomes(11),(12),(13),(14).The importance of waist circumference in predicting health risk is thought to be due to the relationship between waist circumference and intra-abdominal fat(15),(16),(17),(18),(19),(20).We now know that the presence of intra-abdominal visceral fat is associated with a poorer outcome in that patients are prone to develop metabolic syndrome and insulin resistance(21).We have yet to devise a more accurate measurement on visceral fat and at present limited to using waist circumference measurements.
Although doctors are generally good at BMI estimation, we found that in estimating overweight patients’ BMI, close to 30% were wrongly estimated as having normal BMI. Next to the obese, these groups of patients are likely to have metabolic abnormalities and increased cardiovascular risk. If actual measurement of BMI is not routinely done, we may neglect patients who would benefit from intervention. A simple, short counseling during the outpatient visit with emphasis on weight loss, the need to increase their daily activity levels and the morbidity related to being overweight may be all that is needed to improve the population health in general. Further intervention may include referrals to hospital or community dieticians and prescribed exercise programmes. These intervention tools already exist in the healthcare system and could be accessed readily.
The nature of our study design exposes it to several potential selection and measurement biases. Future studies should include patients of differing ages and socioeconomic background. Additionally, clinicians of differing appointments from various different specialties should be included to obtain a more applicable result. A measure of diagnostic efficacy should also be employed to further assess the value of clinical measurement and therapeutic intervention.
Conclusion:
The appropriateness of visual scoring of markers of obesity by doctors is flawed and limited to the obese individuals. True anthropometric measurements would avoid misdiagnosing overweight individuals as normals. We can conclude that patients’ own estimation of weight is unreliable and that they are unaware of the impact of high abdominal fat deposition on cardiovascular outcome. The latter should be addressed in consultations by both hospital physicians and general practitioners. Further emphasis and education in schools and awareness campaigns should also advocate this emerging cardiovascular risk factor.
A 67 year old Caucasian male presented to our institution with a one day history of uncontrollable movements. The patient was being evaluated by a psychiatrist, neurologist and a neuro-opthalmologist for a three month history of severe anxiety, gait instability and palinopsia, respectively. The patient had progressively worsened over the prior two weeks and at the time of presentation reported visual hallucinations with increased confusion. His involuntary movements escalated to the point where it appeared that he was having seizures.
His medical history was significant for gouty arthritis, hypertension, and major depression. His surgical history was notable for an open reduction and internal fixation of his left hip 6 years prior. There was no history of any blood or blood product transfusion. He was an insurance executive and did not have significant occupational exposures. His social and family history was unremarkable. His medications upon arrival included captopril, atenolol, bupropion, lamotrigine, clonazepam, folic acid, and ibuprofen.
On admission he was arousable, well nourished, afebrile, haemodynamically stable but disorientated. His cardiopulmonary, abdominal and integumentary examinations were unremarkable. Neurological examination was significant for bilateral symmetric hyperreflexia, diffuse cogwheel rigidity without a resting or postural tremor, and multi-focal dysrhythmic generalised myoclonus. No neck tenderness or nuchal rigidity was noted. A CT of the head without contrast in the causality department was negative for haemorrhage or any acute intracranial pathology.
His initial assessment showed confusion, hallucinations, myoclonus with medication induced delirium. Lewy body dementia, occipital lobe epilepsy, peduncular hallucinosis and prion disease were all considered in the differential diagnosis. On admission, laboratory data including a CBC, CMP, serum ammonia level, cardiac enzymes, urinalysis, and coagulation profile were unremarkable. A toxicology screen for illicit drugs and heavy metals was negative. The initial lumbar spinal fluid (CSF) analysis was only notable for mild proteinorachia of 57 mg/dL. Gram stain, India-ink stain, acid-fast bacilli (AFB), bacterial & fungal cultures, as well as PCR for viral nucleic acid (herpes, Varicella-Zoster, Epstein-Barr virus, arboviruses, and cytomegalovirus virus) were all negative. MRI with contrast was remarkable only for periventricular ischemic changes consistent with small vessel disease. The patient’s bupropion and lamotrigine were discontinued upon admission, and his clonazepam was increased with resolution of the myoclonus after 24 hours.
The admission EEG showed diffuse slowing with no epileptiform discharges or triphasic waves. Due to progressive neurologic deterioration, he was followed with serial electroencephalograms. On hospital day 5, he became unresponsive and the subsequent EEG revealed non-convulsive status epilepticus (NCSE). Temporary resolution of his seizures was achieved with lorazepam and pentobarbital infusions. After 3 days of almost complete suppression, the pentobarbital was discontinued without NCSE recurrence. On hospital day 15 the EEG again displayed NCSE. A ketamine drip was added to his drug regimen with only brief improvement. Pentobarbital had been restarted and progressively titrated up to the maximal dose without achieving burst suppression. Despite being on the maximal dose of pentobarbital, ketamine, valproic acid, levetiracetam, and topiramate he continued to display NCSE (Figure 1A).
At this point (hospital day 16), therapeutic hypothermia was initiated and continued for 48 hours. The patient’s core body temperature was maintained between 32-33 °C followed by slow rewarming to normothermia over the following 48 hours. Near complete suppression of epileptiform activity was observed on the EEG (Figure 1B). Ketamine and pentobarbital were successfully weaned off during the following days and phenobarbital was introduced without recurrence of NCSE.
Figure 1A. Electroencephalogram from hospital day 15. Refractory non-convulsive status epilepticus while on ketamine, levetiracetam, valproic acid, topiramate and pentobarbital.
Figure 1B. Electroencephalogram from hospital day 16, after the initiation of therapeutic hypothermia. Suppression of epileptiform activity is observed after treatment with therapeutic hypothermia; ECG artifact persisting.
Figure 1C. Electroencephalogram from hospital day 29, 13 days after treatment with therapeutic hypothermia, illustrating generalised periodic sharp wave discharges with lack of background activity. Occasional triphasic waves are noted consistent with Creutzfeldt-Jakob encephalopathy.
Figure 2. RepeatMRI of the brain on hospital day 21 illustrates asymmetric basal ganglia hyperintensities on diffusion weighted sequences, which are often observed in CJD.
Figure 3. H & E stain of the cerebral cortex with low and high magnification (A & B). Coarse and fine vacuolization with spongiosis (arrows) are demonstrated on H & E and silver stain, respectively (C & D).
The patient had a repeat MRI which showed asymmetric basal ganglia hyper intensity on diffusion weighted imaging sequence consistent with CJD 3 (Figure 2)3. The results of CSF analysis for protein 14-3-3, neuron-specific enolase, and tau protein became available on hospital day 22. Despite controlling the NCSE, the patient remained unresponsive over the course of the following weeks. The EEG pattern changed to generalised periodic sharp waves, 1-2 per second with occasional triphasic waves, and a lack of background activity (Figure 1C). After fully reviewing the results with the family, an open brain biopsy was performed in effort to verify the diagnosis. The biopsy confirmed the diagnosis of spongiform encephalopathy (Figure 3). In light of the findings, withdrawal of care was initiated upon the family’s request and the patient expired on hospital day 42. The patient’s estimated symptomatic clinical course was approximately four and one-half months.
DISCUSSION
Creutzfeldt-Jakob disease is the archetype of prion mediated neurodegenerative disorders. There are 4 types of CJD; sporadic, familial, iatrogenic and variant4. The sporadic type accounts for 85 per cent of all cases of CJD4. The diagnosis of CJD and transmissible spongiform encephalopathy (TSE) can be elusive. The World Health Organisation’s diagnostic criteria for CJD require at least one of the following: (1) Neuropathological confirmation, (2) confirmation of protease-resistant prion protein (PrP) via immunohistochemistry or Western blot, or (3) presence of scrapie-associated fibrils4. However, newer and less invasive means for diagnosis have been explored in recent literature. CSF analysis for protein 14-3-3, tau protein, S100B protein and neuron-specific enolase have demonstrated sensitivities of 93 per cent, 89 per cent, 87 per cent and 78 per cent, respectively5. In addition, the use of MRI fluid-attenuated inversion recovery (FLAIR) and diffusion weighted imaging (DWI) techniques have yielded sensitivities of 91-92 per cent and specificities of 94 -95 per cent respectively, especially when utilised early in the disease state6. In our case, the initial MRI was unremarkable and only the repeated MRI, performed three weeks after admission, revealed basal ganglia signal intensities consistent with CJD.
One of the most studied and well characterised tools used to support the diagnosis of CJD is the EEG. The typical pattern observed in the early stages of CJD is frontal intermittent rhythmic delta activities (FIRDA). As the disease progresses, characteristic periodic sharp wave complexes (PSWC) can be observed, usually 8 to 12 weeks after the onset of symptoms7. However, the reported sensitivity of EEG is relatively low, ranging from 22 to 73 per cent, and is largely dependent of the subtype of CJD8. In our case, the patient presented with NCSE, which is an uncommon presentation of an uncommon disease. In a retrospective review of 1,384 patients with probable or definite CJD, only 0.007 per cent or 10 patients presented with NCSE2. Our patient did not demonstrate EEG findings consistent with CJD until late in his hospital course. Hence, CJD must be considered as a diagnosis in a patient who presents with refractory non-convulsive status epilepticus without overlooking the more common causes9.
The last important observation is the potential utility of therapeutic hypothermia in patients with refractory NCSE. Therapeutic hypothermia has long been known to suppress epileptiform discharge10,11. However, the safety and efficacy have not been broadly studied in human subjects. Corry and colleagues conducted a small study examining the effects of therapeutic hypothermia on 4 patients with refractory status epilepticus. The results were promising in that therapeutic hypothermia was successful in aborting seizure activity in all 4 patients and effectively suppressed seizure activity in 2 of the 4 patients after re-warming12. We were able to observe similar result in achieving temporary resolution of NCSE with therapeutic hypothermia in combination with antiepileptic medication in our patient.
Malaria is caused by obligate intra-erythrocytic protozoa of the genus Plasmodium. Humans can be infected with one (or more) of the following five species: P. falciparum, P. vivax, P. ovale, and P. malariae and P. knowlesi. Plasmodia are transmitted by the bite of an infected female Anopheles mosquito and these patients commonly present with fever, headache, fatigue and musculoskeletal symptoms.
Diagnosis is made by demonstration of the parasite in peripheral blood smear. The thick and thin smears are prepared for identification of malarial parasite and genotype respectively. Rapid diagnosis of malaria can be done by fluorescence microscopy with light microscope and interference filter or by polymerase chain reaction.
We report a complicated case of P. ovale malaria without fever associated with Hepatitis B virus infection, pre-excitation (WPW pattern), and secondary adrenal insufficiency.
Case Report:
A 23 year old African American man presented to the emergency department with headache and dizziness for one week. He had 8/10 throbbing headaches associated with dizziness, nausea and ringing sensation in the ears and also complained of sweating but denied any fever. He had loose, watery bowel movements 3 times a day for a few days and had vomited once 5 days ago. He denied any past medical history or family history. He was a chronic smoker and smoked 1PPD for 8 years and denied alcohol or drug use. He had travelled to Africa 9 months before presentation and had stayed in Senegal for 1 month though he did not have any illnesses during or after returning from Africa.
On examination: T: 97.6, HR: 115/min, BP: 105/50, no orthostasis, SPO2: 100% in room air and RR: 18/min. Head, neck and throat examinations were normal and respiratory and cardiovascular system examinations were unremarkable except for tachycardia. Abdominal examination revealed no organomegaly and his CNS examination was unremarkable.
Laboratory examination revealed: WBC: 6.4, Hb: 14.4 and Hct: 41.3, Platelets: 43, N: 83.2, L: 7.4, M: 9.3, B: 0.1. His serum chemistry was normal except for a creatinine of 1.3 (BUN 14) and albumin of 2.6 (total protein 5.7). A pre-excitation (WPW Pattern) was seen on ECG and head CT and Chest X-ray were normal.
He was admitted to the telemetry unit to monitor for arrhythmia. Peripheral blood smear (PBS) was sent because of thrombocytopenia and mild renal failure and revealed malarial parasites later identified as P. ovale (Pic. 1 and 2).
He was treated with Malarone; yet after 2 days of treatment, he was still complaining of headache, nausea and dizziness. There were no meningeal signs. His blood pressure readings were low (95/53) and he was orthostatic. His ECG showed sinus tachycardia and did not reveal any arrhythmias or QTc prolongation. His morning serum cortisol was 6.20 and subsequent cosyntropin stimulation test revealed a serum cortisol of 13.40 at one hour after injection. His Baseline ACTH was<1.1 suggesting a secondary adrenal insufficiency. His IGF-1, TSH, FT4, FSH, LH were all within normal limits. His bleeding and coagulation parameters were normal, CD4 was 634(CD4/CD8: 1.46) and rapid oral test for HIV was negative. His Hepatitis B profile was as follows: HBsAg: positive, HBV Core IgM: negative, HBV core IgG: positive, HBeAg: negative, HBeAb: positive, HBV DNA: 1000 copies/ml, Log10 HBV DNA: 3000 copies/ml.
His Blood cultures were negative, his G6PD levels and hemoglobin electrophoresis were normal, haptoglobin was<15 and LDH was 326. MRI of the brain was unremarkable. The abdominal sonogram revealed a normal echo pattern of the liver and spleen and spleen size was 12 cm. The secondary adrenal insufficiency was treated with dexamethasone resulting in gradual improvement of his nausea, vomiting and headache. Furthermore the platelet count improved to 309. Primaquine was prescribed to complete the course of malaria treatment and he was discharged home following 8 days of hospitalization. Unfortunately he did not return for follow up.
Discussion:
Malaria continues to be a major health problem worldwide. In 2007 the CDC received reports of 1,505 cases of malaria among person in the United States. 326 cases were reported from New York with all but one of these cases being acquired outside of the United States1.
While Plasmodia are primarily transmitted through the bite of an infected female Anopheles mosquito, infections can also occur through exposure to infected blood products (transfusion malaria) or by congenital transmission. In industrialized countries most cases of malaria occur among travellers, immigrants, or military personnel returning from areas endemic for malaria (imported malaria). Exceptionally, local transmission through mosquitoes occurs (indigenous malaria). For non-falciparum malaria the incubation period is usually longer (median 15–16 days) and both P. Vivax and P. Ovale malaria may relapse months or years after exposure due to the presence of hypnozoites in the liver of which the longest reported incubation period for P. vivax being 30 years2.
Malaria without fever has been reported in cases of Plasmodium falciparum malaria in non- immune people3. Hepatitis B infection associated with asymptomatic malaria has been reported in the Brazilian Amazon4. This study was done in P. falciparum and P. vivax infected person with HBV co-infection though not in the P. ovale group. HBV infection leads to increased IFN-gamma levels5,6 which are important for plasmodium clearance in the liver7, in addition to its early importance for malarial clinical immunity8. High levels of IFN gamma, IL6 and TNF alpha are detectable in the blood of malaria patients and in the spleen and liver in the rodents’ model of malaria9,10. These inflammatory cytokines are known to suppress HBV replication in HBV transgenic mice9. This might explain the low levels of HBV viremia in our patient although human studies are required to confirm this finding.
The hypothalamic-pituitary- adrenocortical axis suppression and primary and secondary adrenal insufficiency has been reported in severe falciparum malaria10. In our case, the patient did not have any features to characterize severe malaria, and parasitaemia was <5%. Further, the MRI did not reveal any secondary cause for adrenal insufficiency. This might indicate that patients with malaria are more prone for hypothalamo-pituitary adrenocortical axis dysregulation yet further studies are required to prove this phenomenon in patients without severe malaria.
Cardiac complications after malaria have rarely been reported. In our patient pre-excitation on ECG disappeared after starting antimalarial treatment. Whether WPW pattern and its subsequent disappearance was incidental or caused by malarial infection that improved with treatment could not be determined. Lengthening of the QTc and severe cardiac arrhythmia has been observed, particularly after treatment with halofantrine for chloroquine resistant Plasmodium falciparum malaria11. Post-infectious myocarditis can be associated with cardiac events especially in combination with viral infections12. A case of likely acute coronary syndrome and possible myocarditis was reported after experimental human malaria infection13. To date, except for cardiac arrhythmias that developed after treatment with halofantrine and quinolines, no other arrhythmias has been reported in patients with malaria before treatment.
Transient thrombocytopenia is very common in uncomplicated malaria in semi -immune adults14. A person with a platelet count <150 × 109/l is 4 times more likely to have asymptomatic malarial infection than one with a count ≥150 × 109/l15. In an observational study among 131 patients, patients with involvement of more than one organ system was found to have a lower mean platelet count compared to those with single organ involvement16.
Conclusions:
Our case highlights the need for further studies to understand the multi-organ involvement in patients without severe malaria as well as early recognition of potential complications to prevent mortality and morbidity in this subgroup of patients.
In recent years, increasing attention has focused on the treatment of chronic pain with a considerable number of research and publications about it. At the same time, opioid prescription, use, abuse and death related to the inappropriate use of opioids have significantly increased over the last 10 years. Some reports indicated that there were more than 100 ‘pain clinics’ within a one-mile radius in South Florida, between 2009 and 2010, which led to the birth of new opioid prescription laws in Florida and many other states to restrict the use of opioids. In the face of clinical and social turmoil related to opioid use and abuse, a fundamental question facing each clinician is: are opioids effective and necessary for chronic non-malignant pain?
Chronic low back pain (LBP) is the most common pain condition in pain clinics and most family physician offices, which ‘requires’ chronic use of opioids. Nampiaparampil et al conducted a literature review in 20121 and found only one high-quality study on oral opioid therapy for LBP, which showed significant efficacy in pain relief and patient function. Current consensus believes that there is weak evidence demonstrating favourable effectiveness of opioids compared to placebo in chronic LBP.2Opioids may be considered in the treatment of chronic LBP if a patient fails other treatment modalities such as non-steroidal anti-inflammatory drugs (NSAIDs), antidepressants, physical therapy or steroid injections. Opioids should be avoided if possible, especially in adolescents who are at high risk of opioid overdose, misuse, and addiction. It has been demonstrated that the majority of the population with degenerative disc disease, including a disc herniation have no back pain. A Magnetic Resonance Imaging (MRI) report or film with a disc herniation should not be an automatic ‘passport’ for access to narcotics.
Failed back surgery syndrome (FBSS) is often refractory to most treatment modalities and sometimes very debilitating. There are no well-controlled clinical studies to approve or disapprove the use of opioids in FBSS. Clinical experience suggests oral opioids may be beneficial and necessary to many patients suffering from severe back pain due to FBSS. Intraspinal opioids delivered via implanted pumps may be indicated in those individuals who cannot tolerate oral medications. For elderly patients with severe pain due to spinal stenosis, there is no clinical study to approve or disprove the use of opioids. However, due to the fact that NSAIDs may cause serious side effects in gastrointestinal, hepatic and renal systems, opioid therapy may still be a choice in carefully selected patients.
Most studies for pharmacological treatment of neuropathic pain are conducted with diabetic peripheral neuropathy (DPN) patients. Several randomized clinical controlled studies have demonstrated evidence that some opioids, such as morphine sulphate, tramadol,3 and oxycodone controlled-release,4 are probably effective in reducing pain and should be considered as a treatment of choice (Level B evidence), even though anti-epileptics such as pregabalin should still be used as the first line medication.5
Some studies indicate opioids may be superior to placebo in relieving pain due to acute migraine attacks and Fiorinal with codeine may be effective for tension headache. However there is lack of clinical evidence supporting long-term use of opioids for chronic headaches such as migraine, chronic daily headache, medication overuse headache, or cervicogenic headache. Currently there are large amounts of opioids being prescribed for headaches because of patients' demands. Neuroscience data on the effects of opioids on the brain has raised serious concerns for long-term safety and has provided the basis for the mechanism by which chronic opioid use may induce progression of headache frequency and severity.6 A recent study found chronic opioid use for migraine associated with more severe headache-related disability, symptomology, comorbidities (depression, anxiety, and cardiovascular disease and events), and greater healthcare resource utilization.7
Many patients with fibromyalgia (FM) come into pain clinics to ask for, or even demand, prescriptions for opioids. There is insufficient evidence to support the routine use of opioids in fibromyalgia.8 Recent studies have suggested that central sensitization may play for role in the aetiology of FM. Three central nervous system (CNS) agents (pregabalin, duloxetine and milnacipran) have been approved by United States Food and Drug Administration (US FDA) for treatment of FM. However, opioids are still commonly prescribed by many physicians for FM patients by ‘tradition’, sometimes even with the combination of a benzodiazapine and muscles relaxant - Soma. We have observed negative health and psychosocial status in patients using opioids and labeled with FM. Opioids should be avoided whenever possible in FM patients in face of widespread abuse and lack of clinical evidence.9
Adolescents with mild non-malignant chronic pain rarely require long-term opioid therapy.10 Opioids should be avoided if possible in adolescents, who are at high risk of opioid overdose, misuse, and addiction. Patients with adolescents living at home should store their opioid medication safely.
In conclusion, opioids are effective and necessary in certain cases. However, currently no single drug stands out as the best therapy for managing chronic non-malignant pain, and current opioid treatment is not sufficiently evidence-based. More well-designed clinical studies are needed to confirm the clinical efficacy and necessity for using opioids in the treatment of chronic non-malignant pain. Before more evidence becomes available, and in the face of widespread abuse of opioids in society and possible serious behavioural consequences to individual patients, a careful history and physical examination, assessment of aberrant behavior, controlled substance agreement, routine urine drug tests, checking of state drug monitoring system (if available), trials of other treatment modalities, and continuous monitoring of opioid compliance should be the prerequisites before any opioids are prescribed.
Opioid prescriptions should be given as indicated, not as ‘demanded’.
A 69 year old male with hypertension, body mass index 24 kg/m2, neck circumference 16 inches, and moderate COPD, on home oxygen, presented to his pulmonary clinic appointment with worsening complaints of fatigue, leg cramps, and intermittent shortness of breath with chest discomfort. A remote, questionable history of syncope five to ten years ago was elicited. His vital signs were: temperature 98.80F, blood pressure 119/76 mmHg, pulse 92/min and regular, and respirations 20/min. Physical exam was significant for crowded oropharynx with a Mallampati score of four, distant breath sounds with a prolonged expiratory phase on lung exam with a normal cardiac exam. Laboratory investigation showed normal complete blood counts, haemoglobin 15 g/dL, and normal chemistries. Compared to his previous studies, a pulmonary function study showed stable parameters with a FEV1 1.47 L (69%), FVC/FEV1 ratio 0.44 (62%), and a DLCO/alveolar volume ratio of 2.12 (49%). A room air arterial blood gas revealed pH 7.41, PCO2 44 mmHg, and PO2 61 mmHg, with 92% oxygen saturation. A six minute treadmill exercise test performed to assess the need for supplemental oxygen showed that he required supplemental oxygen at 1L/min via nasal cannula to eliminate hypoxemia during exercise. His chest radiograph was significant for hyperinflation and prominence of interstitial markings. A high resolution computed tomography of the chest demonstrated severe centrilobular and panacinar emphysema only. A baseline electrocardiogram (EKG) showed normal sinus rhythm with an old anterior wall infarct (Figure 1). Echocardiography of the heart revealed a normal left ventricle with an ejection fraction of 65%. Right ventricular systolic function was normal although elevated mean pulmonary arterial pressure of 55 mmHg was noted. A diagnostic polysomnogram performed for evaluation of daytime fatigue and snoring at night revealed mild OSA with an AHI of 6/hr. with sleep time spent with oxygen saturation below 90% (T-90%) of 19%. The EKG showed normal sinus rhythm. A full overnight polysomnogram for continuous positive airway pressure (CPAP) titration performed for treatment of sleep disordered breathing was sub-optimal, however it demonstrated an apnoea–hypopnea index (AHI) of 28 during REM (rapid eye movement) sleep, and a T-90% of 93%. The associated electrocardiogram showed Wenckebach second degree AV heart block during REM sleep usually near the nadir of oxygen desaturation. On a repeat positive airway pressure titration study, therapy with Bilevel pressures (BPAP) of 18/14 cmH20 corrected the AHI and nocturnal hypoxemia to within normal limits during Non REM (NREM) and REM sleep. His electrocardiogram remained in normal sinus rhythm .A twenty-four hour cardiac holter monitor revealed baseline sinus rhythm and confirmed the presence of second degree AV block of the Wenckebach type. A one month cardiac event recording showed normal sinus rhythm with frequent episodes of second degree AV block. These varied from Type I progressing to Type II with a 2:1 and 3:1 AV block, during sleep. Progression to complete heart block was noted with the longest pause lasting 3.9 seconds during sleep. The patient underwent an electrophysiology study with placement of a dual chamber pacemaker. He was initiated on BIPAP therapy. Subsequently, the patient was seen in clinic with improvements in his intermittent episodes of shortness of breath, fatigue, and daytime sleepiness.
Figure 1- Patient’s baseline EKG, normal sinus rhythm. Figure 2 -Progression to Mobitz Type II block 5:07 am. Figure 3 and 4- Sinus pauses, longest interval 11:07 pm 3.9 seconds (Figure 4).
Discussion
In healthy individuals, especially athletes, bradycardia, Mobitz I AV block, and sinus pauses up to 2 seconds are common during sleep and require no intervention5. Cardiac rhythm is controlled primarily by autonomic tone. NREM sleep is accompanied by an increase in parasympathetic, and a decrease in sympathetic, tone. REM sleep is associated with decreased parasympathetic tone and variable sympathetic tone. Bradyarrhythmias in patients with OSA are related to the apnoeic episodes and over 80% are found during REM sleep. During these periods of low oxygen supply, increased vagal activity to the heart resulting in bradyarrhythmias may actually be cardioprotective by decreasing myocardial oxygen demand. This may be important in patients with underlying coronary heart disease.
Some studies have found that Mobitz I AV block may not be benign. Shaw6 et al studied 147 patients with isolated chronic Mobitz I AV block. They inserted pacemakers in 90 patients, 74 patients were symptomatic and 16 patients received a pacemaker for prophylaxis. Outcome data included five-year survival, deterioration of conduction to higher degree AV block, and new onset of various forms of symptomatic bradycardia. They concluded that survival was higher in the paced groups and that risk factors for poor outcomes in patients with Mobitz I included age greater than 45 years old, symptomatic bradycardia, organic heart disease, and the presence of a bundle branch block on EKG.
The Sleep Heart Health Study7 found a higher prevalence of first and second-degree heart block among subjects with sleep-disordered breathing (SDB) than in those without (1.8% vs. 0.3% and 2.2 vs. 0.9%, respectively). Gami et al8observed thatupon review of 112 Minnesota residents who hadundergone diagnostic polysomnography and subsequentlydied suddenly from a cardiac cause, sudden death occurred between the hours of midnight and 6:00 AM in 46% of those with OSA, as compared with 21% of those without OSA. In a study of twenty-three patients with moderate to severe OSA who were each implanted with an insertable loop recorder, about 50% were observed to have frequent episodes of bradycardia and long pauses (complete heart block or sinus arrest) during sleep9. These events showed significant night-to-night intra individual variability and their incidence was under-estimated, only 13%, by conventional short-term EKG Holter recordings.
Physiologic factors predisposing patients with OSA to arrhythmias include alterations in sympathetic and parasympathetic nervous system activity, acidosis, apnoea’s, and arousal2, 10, 11. Some patients with OSA may have an accentuation of the ‘Diving Reflex’. This protective reflex consists of hypoxemia-induced sympathetic augmentation to muscles and vascular beds associated with increased cardiac vagal activity which results in increased brain perfusion, bradycardia and decreased cardiac oxygen demand. In patients with cardiac ischemia, poor lung function (i.e. COPD), or both, it may be difficult to differentiate between these protective OSA-associated Bradyarrhythmias and those which may lead to sudden death. It has been well established that patients with COPD are at higher risk for cardiovascular morbidity12 and arrythmias13. Fletcher14 and colleagues reported that the effects of oxygen supplementation on AHI, hypercapnea and supraventricular arrhythmias in patients with COPD and OSA were variable. Out of twenty obese men with COPD studied, in most patients oxygen eliminated the bradycardia observed during obstructive apnoea’s and eliminated AV block in two patients. In some patients supplemental oxygen worsened end-apnoea respiratory acidosis however this did not increase ventricular arrhythmias.
CPAP therapy has been demonstrated to significantly reduce sleep–related Bradyarrhythmias, sinus pauses, and the increased risk for cardiac death 9, 15. Despite this, in certain situations placement of a pacemaker may be required. These include persistent life-threatening arrhythmias present in patients with severe OSAS on CPAP, arrhythmias in patients who are non-compliant with CPAP, and in patients who may have persistent sympathovagal imbalance and hemodynamic fluctuations resulting in daytime bradyarrhythmias16.
Our case is interesting since it highlights the importance of recognizing the association between OSA, COPD, and life-threatening cardiac arrhythmias. Primary care providers should note the possible association of OSA-associated bradyarrhythmias with life-threatening Type II bradyarrhythmias and pauses. Since bradyarrhythmias related to OSA are relieved by CPAP, one option would be to treat with CPAP and observe for the elimination of these arrhythmias using a 24hour holter or event recorder17. Compliance with CPAP is variable and if life-threatening bradycardia is present, placement of a permanent pacemaker may be preferred18.
Our patient is unusual because most studies showing a correlation with the severity of OSA and magnitude of bradycardia have included overweight patients without COPD19. This patient’s electrocardiogram revealed a Type II AV block at 5am (Figure 2). This is within the overnight time frame where patients with OSA have been observed to have an increased incidence of sudden death. Figures 3 and 4 show significant sinus pauses. In selected cases where patients have significant co-morbidities (i.e. severe COPD with OSA), in addition to treatment with positive airway pressure, electrophysiological investigation with placement of a permanent pacemaker may be warranted.
Even though it is commonly seen in Graves' disease, TPP is not related to the etiology, severity, and duration of thyrotoxicosis. 1
The pathogenesis of hypokalaemic periodic paralysis in certain populations with thyrotoxicosis is unclear. Transcellular distribution of potassium is maintained by the Na+/K+–ATPase activity in the cell membrane, and it is mainly influenced by the action of insulin and beta-adrenergic catecholamines.2 Hypokalemia in TPP results from an intracellular shift of potassium and not total body depletion. It has been shown that the Na+/K+–ATPase activity in platelets and muscles is significantly higher in patients with TPP.3 Hyperthyroidism may result in a hyperadrenergic state, which may lead to the activation of the Na+/K+–ATPase pump and result in cellular uptake of potassium.2, 4, 5 Thyroid hormones may also directly stimulate Na+/K+– ATPase activity and increase the number and sensitivity of beta receptors.2, 6 Patients with TPP have been found to have hyperinsulinemia during episodes of paralysis. This may explain the attacks after high-carbohydrate meals.7
CASE REPORT:
A 19 year old male patient presented to our emergency room with sudden onset weakness of lower limbs. He was not able to stand or walk. Power of 0/5 in both lower limbs and 3/5 in upper limbs was noticed on examination. Routine investigations revealed to have severe hypokalemia with a serum potassium of 1.6 meq/l (normal range 3.5-5.0 meq/l), a serum phosphorus level of 3.4 mg/dl (normal range 3-4.5 mg/dl) and mild hypomagnesemia with serum magnesium level of 1.5mg/dl (normal range 1.8-3.0 mg/dl). ECG showed hypokalemic changes with prolonged PR interval, increased P-wave amplitude and widened QRS complexes. He was managed on intravenous as well oral potassium and history revealed weight loss, increased appetite and tremors from past 4 months. He had a multinodular goiter and radioactive iodine uptake scan (Iodine 131) showed a toxic nodule (Toxic nodule shows increased iodine uptake while the rest of the gland is suppressed) with no exophthalmos, sensory or cranial nerve deficits. Thyroid function tests revealed thyrotoxicosis with free T4 of 4.3ng/dl (normal range 0.8-1.8ng/dl), T3 of 279 ng/dl (normal range = 60 - 181 ng/dl) and a TSH level of <0.15milliunits/L (normal range = 0.3 - 4 milliunits/L). He was managed on intravenous potassium & propanolol. The patient showed dramatic improvement of his symptoms. The patient was discharged home on carbamazole with the diagnosis of TPP secondary to toxic nodular goiter.
In this case there was a significant family history as one of his elder brother had a sudden death (cause not known) and his mother was primary hypothyroid on levothyroxin replacement therapy.
DISCUSSION :
TPP is seen most commonly in Asian populations, with an incidence of approximately 2% in patients with thyrotoxicosis of any cause.1,8,9,10 The attacks of paralysis have a well-marked seasonal incidence, usually occurring during the warmer months.1 Pathogenesis of hypokalaemia has been explained by some authors to be due to an intracellular shift of body potassium, which is catecholamine mediated.11,12 Shizume and his group studied total exchangeable potassium which revealed that patients with thyrotoxic periodic paralysis were not significantly different from controls when the value was related to lean body mass.11 The paralytic symptoms and signs improve as the potassium returns from the intracellular space back into the extracellular space.13 The diurnal variation in potassium movement where there is nocturnal potassium influx into skeletal muscle would explain the tendency for thyrotoxic periodic paralysis to occur at night.14 Hypophosphataemia and hypomagnesaemia are also known to occur in association with thyrotoxic periodic paralysis.14,15,16,17,18 The correction of hypophosphataemia without phosphate administration supports the possibility of intracellular shift of phosphate.16 Electrocardiographic findings supportive of a diagnosis of TPP rather than sporadic or familial periodic paralysis are sinus tachycardia, elevated QRS voltage and first-degree AV block (sensitivity 97%, specificity 65%).20 In addition to ST-segment depression, T-wave flattening or inversion and the presence of U waves are typical of hypokalaemia.
The management is to deal with the acute attack as well as treatment of the underlying condition to prevent future attacks. Rapid administration of oral or intravenous potassium chloride can abort an attack and prevent cardiovascular and respiratory complications.4 A small dose of potassium is the treatment of choice for facilitating recovery and reducing rebound hyperkalaemia due to release of potassium and phosphate from the cells on recovery.1,2,3 Rebound hyperkalaemia occurred in approximately 40% of patients with TPP, especially if they received >90 mmol of potassium chloride within the first 24 hours.4 Another mode of treatment is to give propranolol, a nonselective b-blocker, which prevents the intracellular shift of potassium and phosphate by blunting the hyperadrenergic stimulation of Na+/K+–ATPase.20 Hence, initial therapy for stable TPP should include propranolol.21,22,23 The definitive therapy for TPP includes treatment of hyperthyroidism with antithyroid medications, surgical thyroidectomy, or radioiodine therapy.
Normal sleep is divided into Non-REM and REM. REM occurs every 90-120 minutes during adult sleep throughout the night with each period of REM progressing in length such that the REM periods in the early morning hours are the longest and may last from 30-60 minutes. Overall, REM accounts for 20-25% of the sleep time but is weighted toward the second half of the night. During REM sleep with polysomnography monitoring one observes a low voltage mixed frequency amplitude EEG and low voltage EMG in the chin associated with intermittent bursts of rapid eye movements. During the periods of REM breathing becomes irregular, blood pressure rises and the heart rate also increases due to excess adrenergic activity. The brain is highly active during REM and the electrical activity recorded in the brain by EEG during REM sleep is similar to that of wakefulness.
Parasomnias are undesirable, unexpected, abnormal behavioral phenomena that occur during sleep. There are three broad categories in parasomnias. They are
Disorders of Arousal (from Non-REM sleep)
Parasomnias usually associated with REM sleep, and
Other parasomnias which also includes secondary type of parasomnias.
RBD is the only parasomnia which requires polysomnographic testing as part of the essential diagnostic criteria.
Definition of RBD
“RBD is characterized by the intermittent loss of REM sleep electromyographic (EMG) atonia and by the appearance of elaborate motor activity associated with dream mentation” (ICSD-2).1 These motor phenomena may be complex and highly integrated and often are associated with emotionally charged utterances and physically violent or vigorous activities. RBD was first recognized and described by Schenck CH et al. in 1986.2 This diagnosis was first incorporated in the International Classification of Sleep Disorders (ICSD) in 1990. (American Academy of Sleep Medicine)
A defining feature of normal REM sleep is active paralysis of all somatic musculature (sparing the diaphragm to permit ventilation). This result in diffuse hypotonia of the skeletal muscles inhibiting the enactment of dreams associated with REM sleep. In RBD there is an intermittent loss of muscle atonia during REM sleep that can be objectively measured with EMG as intense phasic motor activity (figure 1 and 2).
Figure 1
Figure 2
This loss of inhibition often precedes the complex motor behaviors during REM sleep. Additionally, RBD patients will report that their dream content is often very violent or vigorous dream enacting behaviors include talking, yelling, punching, kicking, sitting, jumping from bed, arm flailing and grabbing etc. and most often the sufferer will upon waking from the dream immediately report a clear memory of the dream which coincides very well with the high amplitude violent defensive activity witnessed. This complex motor activity may result in a serious injury to the dreamer or bed partner that then prompts the evaluation.
Prevalence
The Prevalence of RBD is about 0.5% in general population.1, 3 RBD preferentially affect elderly men (in 6th and 7th decade) with ratio of women to men being 1 to 9.4 The mean age of disease onset is 60.9 years and at diagnosis is 64.4 years.5 RBD was reported in an 18 year old female with Juvenile Parkinson disease,6 so age and gender are not absolute criteria.
In Parkinson disease (PD) the reported prevalence ranges from 13-50%,7, 14-19 LewyBody Dementia (DLB) 95%,8 and Multiple System Atrophy (MSA) 90 %.9 The presence of RBD is a major diagnostic criterion for MSA. RBD has been reported in Juvenile Parkinson disease, and pure autonomic failure10-12 all neurodegenerative disorders are synucleinopathies.13
Physiology
The neurons of locus coeruleus, raphe nuclei, tuberomammillary nucleus, pedunculopontine nucleus, laterodorsal tegmental area and the perifornical area are firing at a high rate, and cause arousal by activating the cerebral cortex. During REM sleep, the aforementioned excitatory areas fall silent with the exception of the pedunculopontine nucleus and laterodorsal tegmental areas. These regions project to the thalamus and activate the cortex during REM sleep. This cortical activation is associated with dreaming in REM. Descending excitatory fibers from the pedunculopontine nucleus and laterodorsal tegmental area innervate the medial medulla, which then sends inhibitory projections to motor neurons producing the skeletal muscle atonia of REM sleep.20-21
There are two distinct neural systems which collaborate in the “paralysis” of normal REM sleep, one is mediated through the active inhibition by neurons in the nucleus reticularis magnocellularis in the medulla via the ventrolateral reticulospinal tract synapsing on the spinal motor neurons and the other system suppresses locomotor activity and is located in pontine region.22
Pathophysiology
REM sleep contains two types of variables, tonic (occurring throughout the REM period), and phasic (occurring intermittently during a REM period). Tonic elements include desynchronized EEG and somatic muscle atonia (sparing the diaphragm). Phasic elements include rapid eye movements, middle ear muscle activity and extremity twitches. The tonic electromyogram suppression of REM sleep is the result of active inhibition of motor activity originating in the perilocus coeruleus region and terminating in the anterior horn cells via the medullary reticularis magnocellularis nucleus.
In RBD, the observed motor activity may result from either impairment of tonic REM muscle atonia or from increase phasic locomotor drive during REM sleep. One mechanism by which RBD results is the disruption in neurotransmission in the brainstem, particularly at the level of the pedunculopontine nucleus.23Pathogenetically, reduced striatal dopaminergic mediation has been found24-25 in those with RBD. Neuroimaging studies support dopaminergic abnormalities.
Types of RBD
RBD can be categorized based on severity:
Mild RBD occurring less than once per month,
Moderate RBD occurring more than once per month but less than once per week, associated with physical discomfort to the patient or bed partner, and
Severe RBD occurring more than once per week, associated with physical injury to patient or bed partner.
RBD can be categorized based on duration:
Acute presenting with one month or less,
Subacute with more than one month but less than 6 months,
Chronic with 6 months or more of symptoms prior to presentation.
Acute RBD: In 55 - 60% of patients with RBD the cause is unknown, but in 40 - 45% the RBD is secondary to another condition. Acute onset RBD is almost always induced or exacerbated by medications (especially Tri-Cyclic Antidepressants, Selective Serotonin Reuptake Inhibitors, Mono-Amine Oxidase Inhibitors, Serotonin Norepinephrine Reuptake Inhibitors,26 Mirtazapine, Selegiline, and Biperiden) or during withdrawal of alcohol, barbiturates, benzodiazepine or meprobamate. Selegiline may trigger RBD in patients with Parkinson disease. Cholinergic treatment of Alzheimer’s disease may trigger RBD.
Chronic RBD: The chronic form of RBD was initially thought to be idiopathic; however long term follow up has shown that many eventually exhibit signs and symptoms of a degenerative neurologic disorder. One recent retrospective study of 44 consecutive patients diagnosed with idiopathic RBD demonstrated that 45% (20 patients) subsequently developed a neurodegenerative disorder, most commonly Parkinson disease (PD) or Lewy body dementia, after a mean of 11.5 years from reported symptoms onset and 5.1 years after RBD diagnosis.27
The relationship between RBD and PD is complex and not all persons with RBD develop PD. In one study of 29 men presenting with RBD followed prospectively, the incidence of PD was 38% at 5 years and 65% after 12 years.7, 28, 29 Contrast this with the prevalence of the condition in multiple system atrophy, where RBD is one of the primary symptoms occurring in 90% of cases.9 In cases of RBD, it is absolutely necessary not only to exclude any underlying neurodegenerative disease process but also to monitor for the development of one over time in follow up visits.
Clinical manifestations
Sufferers of RBD usually present to the doctor with complaints of sleep related injury or fear of injury as a result of dramatic violent, potentially dangerous motor activity during sleep. 96% of patients reporting harm to themselves or their bed partner. Behaviors during dreaming described include talking, yelling, swearing, grabbing, punching, kicking, jumping or running out of the bed. One clinical clue of the source of the sleep related injury is the timing of the behaviors. Because RBD occurs during REM sleep, it typically appears at least 90 minutes after falling asleep and is most often noted during the second half of the night when REM sleep is more abundant.
One fourth of subjects who develop RBD have prodromal symptoms several years prior to the diagnosis. These symptoms may consist of twitching during REM sleep but may also include other types of simple motor movements and sleep talking or yelling.30-31 Day time somnolence and fatigue are rare because gross sleep architecture and the sleep-wake cycle remain largely normal.
RBD in other neurological disorders and Narcolepsy:
RBD has also been reported in other neurologic diseases such as Multiple Sclerosis, vascular encephalopathies, ischemic brain stem lesions, brain stem tumors, Guillain-Barre syndrome, mitochondrial encephalopathy, normal pressure hydrocephalus, subdural hemorrhage, and Tourette’s syndrome. In most of these there is likely a lesion affecting the primary regulatory centers for REM atonia.
RBD is particularly frequent in Narcolepsy. One study found 36% pts with Narcolepsy had symptoms suggestive of RBD. Unlike idiopathic RBD, women with narcolepsy are as likely to have RBD as men, and the mean age was found to be 41 years.32 While the mechanism allowing for RBD is not understood in this population, narcolepsy is considered a disorder of REM state disassociation. Cataplexy is paralysis of skeletal muscles in the setting of wakefulness and often is triggered by strong emotions such as humor. In narcoleptics who regularly experienced cataplexy, 68% reported RBD symptoms, compared to 14% of those who never or rarely experienced cataplexy.32-33 There is evidence of a profound loss of hypocretin in the hypothalamus of the narcoleptics with cataplexy and this may be a link that needs further investigation in the understanding of the mechanism of RBD in Narcolepsy with cataplexy. It is prudent to follow Narcoleptics and questioned about symptoms of RBD and treated accordingly, especially those with cataplexy and other associated symptoms.
Diagnostic criteria for REM Behavior Disorder(ICSD-2: ICD-9 code: 327.42)1
A. Presence of REM sleep without Atonia: the EMG finding of excessive amounts of sustained or intermittent elevation of submental EMG tone or excessive phasic submental or (upper or lower) limb EMG twitching (figure 1 and 2).
B. At least one of the following is present:
i. Sleep related injurious, potentially injurious, or disruptive behaviors by history
ii. Abnormal REM sleep behaviors documented during polysomnographic monitoring
C. Absence of EEG epileptiform activity during REM sleep unless RBD can be clearly distinguished from any concurrent REM sleep-related seizure disorder.
D. The sleep disturbance is not better explained by another sleep disorder, medical or neurologic disorder, mental disorder, medication use, or substance use disorder.
Differential diagnosis
Several sleep disorders causing behaviors in sleep can be considered in the differential diagnosis, such as sleep walking (somnambulism), sleep terrors, nocturnal seizures, nightmares, psychogenic dissociative states, post-traumatic stress disorder, nocturnal panic disorder, delirium and malingering. RBD may be triggered by sleep apnea and has been described as triggered by nocturnal gastroesophageal reflux disease.
Evaluation and Diagnosis
Detailed history of the sleep wake complaints
Information from a bed partner is most valuable
Thorough medical, neurological, and psychiatric history and examination
Screening for alcohol and substance use
Review of all medications
PSG (mandatory): The polysomnographic study should be more extensive, with an expanded EEG montage, monitors for movements of all four extremities, continuous technologist observation and continuous video recording with good sound and visual quality to allow capture of any sleep related behaviors
Multiple Sleep Latency Test (MSLT): Only recommended in the setting of suspected coexisting Narcolepsy
Brain imaging (CT or MRI) is mandatory if there is suspicion of underlying neurodegenerative disease.
Management
RBD may have legal consequences or can be associated with substantial relationship strain; therefore accurate diagnosis and adequate treatment is important, which includes non-pharmacological and pharmacological management.
Non-pharmacological management: Acute form appears to be self-limited following discontinuation of the offending medication or completion of withdrawal treatment. For chronic forms, protective measures during sleep are warranted to minimize the risks for injury to patient and bed partner. These patients are at fall risk due to physical limitations and use of medications. Protective measure such as removing bed stands, bedposts, low dressers and applying heavy curtains to windows. In extreme cases, placing the mattress on the floor to prevent falls from the bed has been successful.
Pharmacological management: Clonazepam is highly effective in treatment and it is the drug of choice. A very low dose will resolve symptoms in 87 to 90% of patients.4, 5, 7-34 Recommended treatment is 0.5 mg Clonazepam 30 minutes prior to bed time and for more than 90% of patients this dose remains effective without tachyphylaxis. In the setting of breakthrough symptoms the dose can be slowly titrated up to 2.0 mg. The mechanism of action is not well understood but clonazepam appears to decrease REM sleep phasic activity but has no effect on REM sleep atonia.35
Melatonin is also effective and can be used as monotherapy or in conjunction with clonazepam. The suggested dose is 3 to 12 mg at bed time. Pramipexole may also be effective36-38 and suggested for use when clonazepam is contraindicated or ineffective. It is interesting to note that during holidays from the drug, the RBD can take several weeks to recur. Management of patients with concomitant disorder like narcolepsy, depression, dementia, Parkinson disease and Parkinsonism can be very challenging, because medications such as SSRIs, selegiline and cholinergic medications used to treat these disorders, can cause or exacerbate RBD. RBD associated with Narcolepsy, clonazepam is usually added in management and it is fairly effective.
Follow-up
Because RBD may occur in association with neurodegenerative disorder, it is important to consult a neurologist for every patient with RBD as early as possible, especially to diagnose and provide care plan for neurodegenerative disorder, which includes but not limited to early diagnosis and management, regular follow up, optimization of management to provide better quality of life and address medico-legal issues.
Prognosis
In acute and idiopathic chronic RBD, the prognosis with treatment is excellent. In the secondary chronic form, prognosis parallels that of the underlying neurologic disorder. Treatment of RBD should be continued indefinitely, as violent behaviors and nightmares promptly reoccur with discontinuation of medication in almost all patients.
Conclusions
RBD and neurodegenerative diseases are closely interconnected. RBD often antedates the development of a neurodegenerative disorder; diagnosis of idiopathic RBD portends a risk of greater than 45% for future development of a clinically defined neurodegenerative disease. Once identified, close follow-up of patients with idiopathic RBD could enable early detection of neurodegenerative diseases. Treatment for RBD is available and effective for the vast majority of cases.
Key Points
Early diagnosis of RBD is of paramount importance
Polysomnogram is an essential diagnostic element
Effective treatment is available
Early treatment is essential in preventing injuries to patient and bed partner
Apparent idiopathic form may precede development of Neurodegenerative disorder by decades
Saliva is the watery and usually frothy substance produced in and secreted from the three paired major salivary (parotid, submandibular and sublingual) glands and several hundred minor salivary glands, composed mostly of water, but also includes electrolytes, mucus, antibacterial compounds, and various enzymes. Healthy persons are estimated to produce 0.75 to 1.5 liters of saliva per day. At least 90% of the daily salivary production comes from the major salivary glands while the minor salivary glands produce about 10%. On stimulation (olfactory, tactile or gustatory), salivary flow increases five fold, with the parotid glands providing the preponderance of saliva.1
Saliva is a major protector of the tissues and organs of the mouth. In its absence both the hard and soft tissues of the oral cavity may be severely damaged, with an increase in ulceration, infections, such as candidiasis, and dental decay. Saliva is composed of serous part (alpha amylase) and a mucus component, which acts as a lubricant. It is saturated with calcium and phosphate and is necessary for maintaining healthy teeth. The bicarbonate content of saliva enables it to buffer and produce the condition necessary for the digestion of plaque which holds acids in contact with the teeth. Moreover, saliva helps with bolus formation and lubricates the throat for the easy passage of food. The organic and inorganic components of salivary secretion have got a protective potential. They act as barrier to irritants and a means of removing cellular and bacterial debris. Saliva contains various components involved in defence against bacterial and viral invasion, including mucins, lipids, secretory immunoglobulins, lysozymes, lactoferrin, salivary peroxidise, and myeloperoxidase. Salivary pH is about 6-7, favouring digestive action of salivary enzyme, alpha amylase, devoted to starch digestion.
Salivary glands are innervated by the parasympathetic and sympathetic nervous system. Parasympathetic postganglionic cholinergic nerve fibers supply cells of both the secretory end-piece and ducts and stimulate the rate of salivary secretion, inducing the formation of large amounts of a low-protein, serous saliva. Sympathetic stimulation promotes saliva flow through muscle contractions at salivary ducts. In this regard both parasympathetic and sympathetic stimuli result in an increase in salivary gland secretions. The sympathetic nervous system also affects salivary gland secretions indirectly by innervating the blood vessels that supply the glands.
Table 1: Functions of saliva
Digestion and swallowing Initial process of food digestion Lubrication of mouth, teeth, tongue and food boluses Tasting food Amylase- digestion of starch Disinfectant and protective role Effective cleaning agent Oral homeostasis Protect teeth decay, dental health and oral odour Bacteriostatic and bacteriocidal properties Regulate oral pH Speaking Lubricates tongue and oral cavity
Drooling (also known as driveling, ptyalism, sialorrhea, or slobbering) is when saliva flows outside the mouth, defined as “saliva beyond the margin of the lip”. This condition is normal in infants but usually stops by 15 to 18 months of age. Sialorrhea after four years of age generally is considered to be pathologic. The prevalence of drooling of saliva in the chronic neurological patients is high, with impairment of social integration and difficulties to perform oral motor activities during eating and speech, with repercussion in quality of lifeDrooling occurs in about one in two patients affected with motor neuron disease and one in five needs continuous saliva elimination7, its prevalence is about 70% in Parkinson disease8, and between 10 to 80% in patients with cerebral palsy9.
Pathophysiology
Pathophysiology of drooling is multifactorial. It is generally caused by conditions resulting in
Excess production of saliva- due to local or systemic causes (table 2)
Inability to retain saliva within the mouth- poor head control, constant open mouth, poor lip control, disorganized tongue mobility, decreased tactile sensation, macroglossia, dental malocclusion, nasal obstruction.
Problems with swallowing- resulting in excess pooling of saliva in the anterior portion of the oral cavity e.g. lack of awareness of the build-up of saliva in the mouth, infrequent swallowing, and inefficient swallowing.
Drooling is mainly due to neurological disturbance and less frequently to hyper salivation.Under normal circumstances, persons are able to compensate for increased salivation by swallowing. However, sensory dysfunction may decrease a person’s ability to recognize drooling and anatomic or motor dysfunction of swallowing may impede the ability to manage increased secretion.
Depending on duration of drooling, it can be classified as acute e.g. during infections (epiglottitis, peritonsilar abscess) or chronicneurological causes.
Symptoms
Drooling of saliva can affect patient and/or their carers quality of life and it is important to assess the rate and severity of symptoms and its impact on their life.
Table 3 Effect of untreated Drooling of saliva
Physical
Psychological
Perioral chapping (skin cracking) Maceration with secondary infection Dehydration Foul odour Aspiration/ pneumonia Speech disturbance Interference with feeding
Isolation Barriers to education (damage to books or electronic devices) Increased dependency and level/intensity of care Damage to electronic devices Decreased self esteem Difficult social interaction
Assessment
Assessment of the severity of drooling and its impact on quality of life for the patient and their carers help to establish a prognosis and to decide the therapeutic regimen. A variety of subjective and objective methods for assessment of sialorrhoea have been described3.
History (from patient and carers)
Establish possible cause, severity, complications and possibility of improvement, age and mental status of patient, chronicity of problems, associated neurological conditions, timing, provoking factors, estimation of quantity of saliva – use of bibs, clothing changing required/ day and impact on the day today life (patient/carer)
Physical examination
Evaluate level of alertness, emotional state, hydration status, hunger, head posture
Examination of oral cavity- sores on the lip or chin, dental problems, tongue control, swallowing ability, nasal airway obstruction, decreased intraoral sensitivity, assessment of health status of teeth, gum, oral mucosa, tonsils, anatomical closure of oral cavity, tongue size and movement, jaw stability. Assessment of swallowing
Assess severity and frequency of drooling (as per table 4)
Investigation
Lateral neck x ray (in peritonsilar abscess)
Ultrasound to diagnose local abscess
Barium swallow to diagnose swallowing difficulties
Audiogram- to rule out conductive deafness associated with oropharyngeal conditions
Salivary gland scan- to determine functional status
Table 4 : System for assessment of frequency and severity of drooling
Drooling severity
Points
Dry (never drools)
1
Mild (wet lips only)
2
Moderate (wet lips and chins)
3
Severe (clothing becomes damp)
4
Profuse (clothing, hands, tray, object become wet)
5
Frequency
Points
Never drools
1
Occasionally drools
2
Frequency drools
3
Constantly drools
4
Other methods of assessing salivary production and drooling
1) 1- 10 visual analogue scale (where 1 is best possible and 10 is worst possible situation)
2) Counting number of standard sized paper handkerchiefs used during the day
3) Measure saliva collected in cups strapped to chin
4) Inserting pieces of gauze with a known weight into oral cavity for a specific period of time and then re-measuring weight and calculating the difference between the dry and wet weights.
6) Salivary duct canulation 12 and measuring saliva production.
Management
Drooling of saliva, a challenging condition, is better managed with a multidisciplinary team approach. The team includes primary care physician, speech therapist, occupational therapist, dentist, orthodontist, otolaryngologist, paediatrician and neurologist. After initial assessment, a management plan can be made with the patient. The person/ carer should understand the goal of treating drooling is a reduction in excessive salivary flow, while maintaining a moist and healthy oral cavity. Avoidance of xerostomia (dry mouth) is important.
There are two main approaches
Non invasive modalities e.g. oral motor therapy, pharmacological therapy
Invasive modalities e.g. surgery and radiotherapy
No single approach is totally effective and treatment is usually a combination of these techniques. The first step in management of drooling is correction of reversible causes. Less invasive and reversible methods, namely oral motor therapy and medication are usually implemented before surgery is undertaken5
Non invasive modalities
Positioningprior to implementation of any therapy, it is essential to look at the position of the patient. When seated, a person should be fully supported and comfortable. Good posture with proper trunk and head control provides the basis for improving oral control of drooling and swallowing.
Eating and drinking skills-drooling can be exacerbated by pooreating skills. Special attention and developing better techniques in lip closure, tongue movement and swallowing may lead to improvements of some extent. Acidic fruits and alcohol stimulate further saliva production, so avoiding them will help to control drooling10
Oral facial facilitation - this technique will help to improve oral motor control, sensory awareness and frequency of swallowing.Scott and staios et al 18 noted improvement in drooling in patients with both hyper and hypo tonic muscles using this technique. This includes different techniques normally undertaken by speech therapist, which improves muscle tone and saliva control. Most studies show short term benefit with little benefit in long run. This technique can be practiced easily, with no side effects and can be ceased if no benefits noted.
a) Icing – effect usually last up to 5-30 minutes. Improves tone, swallow reflex.
b) Brushing- as effect can be seen up to 20- 30 minutes, suggested to undertake before meals.
c) Vibration- improves tone in high tone muscles
d) Manipulation – like tapping, stroking, patting, firm pressure directly to muscles using fingertips known to improve oral awareness.
e) Oral motor sensory exercise - includes lip and tongue exercises.
Speech therapy-speech therapy should be started early to obtain good results. The goal is to improve jaw stability and closure, to increase tongue mobility, strength and positioning, to improve lip closure (especially during swallowing) and to decrease nasal regurgitation during swallowing.
Behaviour therapy-this uses a combination of cueing, overcorrection, and positive and negative reinforcement to help drooling. Suggested behaviours, like swallowing and mouth wiping are encouraged, whereas open mouth and thumb sucking are discouraged. Behavior modification is useful to achieve (1) increased awareness of the mouth and its functions, (2) increased frequency of swallowing, (3) increased swallowing skills. This can be done by family members and friends. Although there is no randomized controlled trial done, over 17 articles published in last 25 years, show promising results and improved quality of life. No reported side effects make behavioural interventions an initial option compared to surgery, botulinum toxin or pharmaceutical management. Behaviour interventions are useful prior and after medical management such as botulinum toxin or surgery.
Oral prosthetic device- variety of prosthetic devices can be beneficial, e.g. chin cup and dental appliances, to achieve mandibular stability, better lip closure, tongue position and swallowing. Cooperation and comfort of the patient is essential for better results.
Pharmacological methods
Systematic review of anticholinergic drugs, show Benztropine, Glycopyrrolate, and Benzhexol Hydrochloride, as being effective in the treatment of drooling. But these drugs have adverse side-effects and none of the drugs been identified as superior.
Hyoscine- The effect of oral anticholinergic drugs has been limited in the treatment of drooling. Transdermal scopolamine (1.5 mg/2.5 cm2) offers advantages. One single application is considered to render a stable serum concentration for 3 days. Transdermal scopolamine has been shown to be very useful in the management of drooling, particularly in patients with neurological or neuropsychiatric disturbances or severe developmental disordersIt releases scopolamine through the skin into the bloodstream.
Glycopyrrolatestudies have shown 70-90% response rates but with a high side effect rate. Approximately 30-35% of patients choose to discontinue due to unacceptable side effects such as excessive dry mouth, urinary retention, decreased sweating, skin flushing, irritability and behavior changes. A study on 38 patients with drooling due to neurological deficits had shown up to a 90% response rateMier et al21 reported Glycopyrrolate to be effective in the control of excessive sialorrhea in children with developmental disabilities. Approximately 20% of children given glycopyrrolate may experience substantial adverse effects, enough to require discontinuation of medication.
Antimuscarinic drugs, such as benzhexol, have also been used, but limited due to their troublesome side effects.
Antireflux Medication: The role of antireflux medication (Ranitidine & Cisapride) in patients with gastro esophageal reflux due to esophageal dysmotility and lower esophageal tone did not show any benefits in a study 21.
Modafinil - One case study noticed decreased drooling in two clients who were using the drug for other reasons, but no further studies have been done.
Alternate medications: (Papaya and Grape seed extract) – Mentioned in literature as being used to dry secretions but no research in to their efficacy has been conducted.
Botulinum toxin It was in 1822 that a German poet and physician, Justinus Kerner, discovered that patients who suffered from botulism complained of severe dryness of mouth which suggested that the toxin causing botulism could be used to treat hypersalivation. However, it was only in the past few years that botulinum toxin type A (BTx-A)has been used for this purpose. BTx-A binds selectively to cholinergic nerve terminals and rapidly attaches to acceptor molecules at the presynaptic nerve surface. This inhibits release of acetylcholine from vesicles, resulting in reduced function of parasympathetic controlled exocrine glands. The blockade though reversible is temporary as new nerve terminals sprout to create new neural connections. Studies have shown that injection of botulinum toxin to parotid and submandibular glands, successfully subsided the symptoms of drooling 30,31. Although there is wide variation in recommended dosage, most studies suggest that about 30- 40 units of BTx-A injected into the parotid and submandibular glands are enough for the symptoms to subside The injection is usually given under ultrasound guidance to avoid damage to underlying vasculature/ nerves. The main side effects from this form of treatment are dysphagia, due to diffusion into nearby bulbar muscles, weak mastication, parotid gland infection, damage to the facial nerve/artery and dental caries.
Patients with neurological disorders who received BTX-A injections showed a statistically significant effect from BTX-A at 1 month post injection, compared with control, this significance was maintained at 6 months. Intrasalivary gland BTX-A was shown to have a greater effect than scopolamine.
The effects of BTx-A are time limited and this varies between individuals.
Invasive modalities
Surgerycan be performed to remove salivary glands, (most surgical procedures focused on parotid and submandibular glands). ligate or reroute salivary gland ducts, or interrupt parasympathetic nerve supply to glands. Wilke, a Canadian plastic surgeon, was the first to propose and carry out parotid duct relocation to the tonsillar fossae to manage drooling in patients with cerebral palsy. One of the best studied procedures, with a large number of patients and long term follow up data, is submandibular duct relocation 32, 33.
Intraductal laser photocoagulation of the bilateral parotid ducts has been developed as a less invasive means of surgical therapy. Early reports have shown some impressive results34.
Overall surgery reducedsalivary flow and drooling can be significantly improved often with immediate results – 3 studies noted that 80 – 89% of participants had an improvement in their control of their saliva. Two studies discussed changes in quality of life. One of these found that 80% of those who participated improved across a number of different measures including receiving affection from others and opportunities for communication and interaction. Most evidence regarding surgical outcomesof sialorrhea management is low quality and heterogeneous. Despitethis, most patients experience a subjective improvement followingsurgical treatment 36.
Radiotherapy - to major salivary glands in doses of 6000 rad or more is effective Side effects which include xerostomia, mucositis, dental caries, osteoradionecrosis, may limit its use.
Key messages
Chronic drooling can pose difficulty in management
Early involvement of Multidisciplinary team is the key.
Combination of approach works better
Always start with noninvasive, reversible, least destructive approach
Surgical and destructive methods should be reserved as the last resort.
Lumbar punctures are commonly performed by both medical and anaesthetic trainees but in different contexts. Medically performed lumbar punctures are often used to confirm a diagnosis (meningitis, subarachnoid haemorrhage) whilst lumbar puncture performed by anaesthetists are usually a precedent to the injection of local anaesthetics into cerebrospinal fluid for spinal anaesthesia. The similarity relies on the fact that both involve the potential for iatrogenic infection into the subarachnoid space. The incidence of iatrogenic infection is very low in both fields; a recent survey by the Royal College of Anaesthetists1 reported an incidence of 8/707 000 whilst there were only approximately 75 cases in the literature after ‘medical’ lumbar puncture.2 However, the consequences of iatrogenic infection can be devastating. It is likely that appropriate infection control measures taken during lumbar puncture would reduce the risk of bacterial contamination. The purpose of the present study is to compare infection control measures taken by anaesthetic and medical staff when performing lumbar puncture.
Method
A survey was constructed online (www.surveymonkey.com) and sent by email to 50 anaesthetic and 50 acute medical trainees in January 2011. All participants were on an anaesthetic or medical training programme and all responses were anonymous. The survey asked whether trainees routinely used the following components of an aseptic technique3 when performing lumbar puncture:
Sterile trolley
Decontaminate hands
Clean patient skin
Apron/gown
Dressing pack
Non-touch technique
Sterile gloves
No ethical approval was sought as the study was voluntary and anonymous.
Results
The overall response rate was 71% (40/50 anaesthetic trainees and 31/50 medical). All anaesthetic trainees routinely used the components of an aseptic technique when performing lumbar puncture. All medical trainees routinely cleaned the skin, decontaminated their hands and used a non-touch technique but only 80.6% used sterile gloves. 61.3% of medical trainees used a sterile trolley, 38.7% used an apron/gown and 77.4% used a dressing pack.
Discussion
This survey shows that adherence to infection control measures differ between anaesthetic and medical trainees when performing lumbar puncture. The anaesthetic trainees have a 100% compliance rate compared to 80% for the medical trainees for all components of the aseptic technique. Both groups routinely cleaned the patient’s skin, decontaminated their hands and used a non-touch technique. However, there were significant differences in the use of other equipment, with fewer medical trainees using sterile gloves, trolleys, aprons and dressing packs.
Although the incidence of iatrogenic infection after lumbar puncture is low, it is important to contribute to this low incidence by adopting an aseptic technique. There may be differences with regards to the risks of iatrogenic infection between anaesthetic and medical trainees. Anaesthetic lumbar punctures involve the injection of a foreign substance (local anaesthesia) into the cerebrospinal fluid and may therefore carry a higher risk. Crucially however, both anaesthetic and medical lumbar punctures involve accessing the subarachnoid space with medical equipment and so the risk is present.
There are many reasons for the differing compliance rates between the two specialties. Firstly, anaesthetic trainees perform lumbar punctures in a dedicated anaesthetic room whilst the presence of ‘procedure/treatment rooms’ is not universal on medical wards. Secondly, anaesthetic trainees will always have a trained assistant present (usually an operating department practitioner, ODP) who can assist with preparing equipment such as dressing trolleys.
The mechanism of iatrogenic infection during lumbar puncture is not completely clear.4 The source of microbial contamination could be external (incomplete aseptic technique, infected equipment) or internal (bacteraemia in the patient); the fact that a common cause of iatrogenic meningitis are viridans streptococcus strains5 (mouth commensals) supports the notion that external factors are relevant and an aseptic technique is important.
It is very likely that improved compliance amongst acute medical trainees would result from a dedicated treatment room on medical wards, but this is likely to involve financial and logistical barriers. The introduction of specific ‘lumbar puncture packs’, which include all necessary equipment (e.g. cleaning solution, aprons, sterile gloves) may reduce the risk of infection; the introduction of a specific pack containing equipment for central venous line insertion reduced colonisation rates from 31 to 12%.6 The presence of trained staff members to assist medical trainees when performing lumbar puncture may assist in improved compliance, similar to the role of an ODP for anaesthetic trainees.
The main limitation of this study is that the sample size is small. However, we feel that this study raises important questions as to why there is a difference in infection control measures taken by anaesthetic and medical trainees; it may be that the environment in which the procedure takes place is crucial and further work on the impact of ‘procedure rooms’ on medical wards is warranted.
Hepatitis B (HB) is a major disease and is a serious global public health problem. About 2 billion people (latest figures so far by WHO) are infected with the hepatitis B virus (HBV) all over the world. Interestingly, rates of new infection and acute disease are highest among adults, but chronic infection is more likely to occur in persons infected as infants or young children, which leads to cirrhosis and hepatocellular carcinoma in later life. More than 350 million persons are reported to have chronic infection globally at present1,2. These chronically infected people are at high risk of death from cirrhosis and liver cancer. This virus kills about 1 million persons each year. For a newborn infant whose mother is positive for both HB surface antigen (HBsAg) and HB e antigen (HBeAg), the risk of chronic HB Virus (HBV) infection is 70% - 90% by the age of 6 months in the absence of post-exposure immunoprophylaxis3.
HB vaccination is the only effective measure to prevent HBV infection and its consequences. Since its introduction in 1982, recommendations for HB vaccination have evolved into a comprehensive strategy to eliminate HBV transmission globally4. In the United States during 1990–2004, the overall incidence of reported acute HB declined by 75%, from 8.5 to 2.1 per 100,000 population. The most dramatic decline occurred in children and adolescents. Incidence among children aged <12 years and adolescents aged 12-19 years declined by 94% from 1.1 to 0.36 and 6.1 to 2.8 per 100,000 population, respectively2,5.
Population of countries with intermediate and high endemicity rates are at high risk of acquiring HB infection. Pakistan lies in an intermediate endemic region with a prevalence of 3-4% in the general population6. WHO has included the HB vaccine in the Expanded Programme on Immunisation (EPI) globally since 1997. Pakistan included the HB vaccination in the EPI in 2004. Primary vaccination consists of 3 intramuscular doses of the HB vaccine. Studies show seroprotection rates of 95% with standard immunisation schedule at 0, 1 and 6 months using a single antigen HB vaccine among infants and children7,8. Almost similar results have been reported with immunisation schedules giving HB injections (either single antigen or in combination vaccines) at 6, 10 and 14 weeks along with other vaccines in the EPI schedule. But various factors like age, gender, genetic and socioenvironmetal influences, are likely to affect seroprotection rates9.So there is need to know actual seroprotection rates in our population where different vaccines, EPI procured and privately procured incorporated in different schedules are used. This study has been conducted to know the real status of seroprotection against HB in our children. Results will help in future policy-making, highlighting our shortcomings, comparing our programme with international standards and moreover augment future confidence in vaccination programmes.
Materials And Methods
This study was conducted at vaccinations centres and paediatrics OPDs (Outpatient Departments) of CMH and MH, Rawalpindi, Pakistan. Children reporting for measles vaccination at vaccination centres at 9 months of age were included. Their vaccination cards were examined and ensured that they had received 3 doses of HB vaccine according to the EPI schedule, duly endorsed in their cards. They included mainly children of soldiers but some civilians also who were invited for EPI vaccination at the MH vaccination centre. Children of officers were similarly included from the CMH vaccination centre and vaccination record was ensured by examining their vaccination cards. Some civilians who received private HB vaccination were included from paediatric OPDs . Some children beyond 9 months and less than 2 years of age who reported for non-febrile minor illnesses in the paediatric OPD at CMH and MH, were also included and their vaccination status was confirmed by examining their vaccination cards.
Inclusion Criteria
1) Male and female children >9 months and <2 years of age.
2) Children who had received 3 doses of HBV according to the EPI schedule at 6,10 and 14 weeks.
3) Children who had a complete record of vaccination- duly endorsed in vaccination cards.
4) Childen who did not have history of any chronic illness.
Exclusion Criteria
1) Children who did not have proper vaccination records endorsed in their vaccination cards.
2) Interval between last dose of HBV and sampling was <1 month.
3) Children suffering from acute illness at time of sampling.
4) Children suffering from chronic illness or on immunosuppressive drugs.
Informed consent for blood sample collection was obtained from the parents or guardians. The study and the informed consent form was approved by the institutional ethical review board. Participants were informed about results of HBs antibody screening. After proper antiseptic measures, blood samples (3.5 ml) were obtained by venepuncture. Autodisabled syringes were used. Collected blood samples were taken in vaccutainers and labelled by identification number and name of child. Samples were immediately transported to the Biochemistery Department of Army Medical College. Samples were kept upright for half an hour and then centrifuged for 10 minutes. Supernatant serum was separated and stored at -20 0C in 1.5 ml eppendorf tubes till the test was performed. Samples were tested using ELISA (DiaSorin S.p.A Italy kit) for detection of anti-HBs antibodies according to manufacturers’ instructions. The diagnostic specificity of this kit is 98.21% (95% confidence interval 97.07-99.00%) and diagnostic sensitivity is 99.11% (95% confidence interval 98.18-99.64%) as claimed by the manufacturer. Anti-HBs antibody enumeration was done after all 3 doses of vaccination (at least 1 month after the last dose was received).
As per WHO standards, anti-HBs antibody titres of >10 IU/L is taken as protective and samples showing antibody titres <10 IU/L were considered as non-protected. Samples having antibody titres >10 IU/L were taken as seroprotected against HB infection. All relevant information was entered in a predesigned data sheet and used accordingly at the time of analysis. Items entered included age, gender, place of vaccination, type of vaccination (private or government procured), number of doses and entitlement status (dependent of military personnel or civilian). The study was conducted from 1st January 2010 to 31st Dec 2010.
Statistical Analysis
Data was analysed using SPSS version 15. Descriptive statistics were used to describe the data, i.e. mean and standard deviation (SD) for quantitative variables, while frequency and percentages were used for qualitative. Quantitative variables were compared through independent samples’ t-test and qualitative variables were compared through the chi-square test between both the groups. A P-value <0.05 was considered as significant.
The mean age of the children was 13.7 months. The overall frequency of children with titres <10 IU/L was 61 (31.4%) while frequency of children with titres >10 IU/L was 133 (68.6%).
Geometric mean titres (GMT) were 85.81 for the seroprotected (>10 IU/L) category.
Results
One hundred and ninety-four children, who had received HB vaccination according to EPI schedule, were tested for anti-HBs titres. Out of them 61 (31.4%) had anti-HBs titres less than 10 IU/L (non-protective level) while 133 (68.6%) had anti-HBs titres above 10 IU/L (protective level) as shown in Figure 1. The GMT of anti-HBs among the individuals having protective levels (> 10 IU/L) was found to be 85.81 IU/L.
Figure 1
Figure 2
Figure 2 shows that anti-HBs titres between 10–100 IU/L was found in 75 (50.4%) children. Twenty-six (19.5%) individuals had titres between 100–200 IU/L. Twenty (14%) children had titres between 20–500 IU/L, 10 (7%) children had titres between 500–1000 IU/L and only 2 (1.5%) children had anti-HBs titres > 1000 IU/L.
One hundred and eighty-four children received vaccination supplied by government sources (Quinevaxem by Novartis) out of which 61 (33.1%) children had anti-HBs titres <10 IU/L (non- protective) and 123 (66.9%) had anti-HBs titres >10 IU/L (protective level). Only 10 children had received vaccination obtained from a private source (Infanrix Hexa by GSK), out of which all 10 (100%) had anti-HBs titres >10 IU/L (protective level). Comparison between the two groups revealed the difference to be significant (P value= 0.028).
One hundred and thirty-two children received vaccination from army health facilities (CMH and MH) out of which 36 (27.3%) had anti-HBs titres < 10 IU/L while 96 (72.7%) had anti-HBs titres >10 IU/L. Sixty-two children were vaccinated at civilian health facilities (health centres or vaccination teams visiting homes). Out of them 25 (40.3%) had anti-HBs titres <10 IU/L while 37 (59.7%) had anti- HBs titres >10 IU/L. The difference was insignificant (P value= 0.068). Gender analysis revealed that in the study group 129 (68.5%) were male children. Out of them 34 (26.4%) had anti-HBs titres <10 IU/L and 95 (73.6%) had anti-HBs titres >10 IU/L. Sixty-five (31.5%) were female children and out of them 27 (41.5%) had anti-HBs titres <10 IU/L while 38 (58.5%) had anti-HBs titres > 10 IU/L. Statistical analysis revealed the difference between males and females was significant (P value= 0.032).
One hundred and twenty-two (62.9%) children were less than 1 year of age. Out of them 37 (30.3%) had anti-HBs titres <10 IU/L and 85 (69.7%) had anti- HBs titres >10 IU/L. Seventy-two (37.1%) children ranged between 1 to 2 years of age. Out of them 24 (33.3%) had anti-HBs titres <10 IU/L while 48 (66.7%) had anti-HBs titres >10 IU/L. On comparison the difference between the two groups was insignificant (P value= 0.663), as shown in Table 1.
Patient characteristics
Anti-HBs titres (< 10 IU/L) (n = 61)
Anti-HBs titres (> 10 IU/L) (n = 133)
P – values
Age groups
0.63 NS
< 1year (n = 122)
37 (30.0%)
85 (69.7%)
> 1year (n = 72)
24 (33.3%)
48 (66.7%)
Gender
0.032
Male (n = 129)
34 (26.4%)
95 (73.6%)
Female (n = 65)
27 (41.5%)
38 (58.5%)
Hospital
0.068 NS
Army (n = 132)
36 (27.3%)
96 (72.7%)
Civilian (n = 62)
25 (40.3%)
37 (59.7%)
Vaccine Type
0.028
Government (n = 184)
61 (33.2%)
123 (66.8%)
Private ( n = 10)
0 (0%)
10 (100%)
Table 1 (NS = Insignificant; * = Significant )
Discussion
HB is a global health problem with variable prevalence in different parts of the world1. Various studies carried out in different parts of Pakistan in different groups of population have shown diverse figures regarding prevalence of HB. However, a figure of 3-4% is accepted as general consensus by and large, thus making Pakistan an area of intermediate endemicity for HB6. Yet when we extrapolate these figures to our population, it is estimated that Pakistan hosts about seven million carriers of HB which is about 5% of the worldwide 350 million carriers of HB10,11.
Age at the time of infection plays the most important role in acquisition of acute or chronic HBV disease. HBV infection acquired in infancy is responsible for a very high risk of chronic liver disease due to HBV in later life12. HB is a preventable disease and fortunately vaccination at birth and during infancy can eradicate the disease globally, if vaccination strategy is effectively implemented13. This can be claimed as the first anti-cancer vaccine which prevents hepatocellular carcinoma in later life.
In Pakistan, the HB vaccine was included in the EPI in 2004, given along with DPT (Diphtheria, Pertussis, Tetanus) at 6, 10 and 14 weeks of age. The vaccine is provided through government health infrastructure to health facilities. Private HB vaccines supplied as a single antigen or in combination vaccines are also available in the market. The efficacy of these recombinant vaccines is claimed to be more than 95% among children and 90% among normal healthy adults14. The immunity of the HB vaccination is directly measured by development of anti-HBs antibodies more than 10 IU/L, which is considered as a protective level15. However, it is estimated that 5–15 % of vaccine recipients may not develop this protective level and remain non-responders due to undermentioned reasons.16 Published studies regarding antibody development in relation to various factors in terms of immunogenicity and seroprotection, show highly varied results. Multiple factors like dose, dosing schedules, sex, storage, site and route of administration, obesity, genetic factors, diabetes mellitus and immunosupression, affect HB antibodies development response17.
Although the HB vaccine was included in the EPI in 2004 in Pakistan, until now no published data showing seroconversion and seroprotection among vaccine recipients of this programme is available on a national level to our knowledge. Our study has revealed that out of 194 children, only 133 (68.6%) had anti-HBs titres in the protective range (>10 IU/L) while 61 (31.4%) did not develop seroprotection. These results are low as compared to other international studies. A study from Bangladesh among EPI vaccinated children shows a seroprotection rate of 92.2%13 while studies from Brazil18 and South Africa19 have separately reported seroprotection rates of 90.0% and 86.6%, respectively. Studies from Pakistan carried out in adults also show seroprotection rates (anti-HBe titres >10 IU/L) of more than 95% in Karachi University students14 and 86% in health care workers of Agha Khan University Hospital20, respectively. However, in these studies the dosing schedule was 0, 1 and 6 months, and participants were adults. These results are consistent with international reports.
The gravity of low seroprotection after HB vaccination is further aggravated when we extrapolate these figures to our overall low vaccination coverage rates of 37.6% to 45% as shown in studies at Peshawar and Karachi respectively21,22. One can imagine a significantly high percentage of individuals vulnerable to HBV infection even after receiving HB vaccine in an extensive national EPI programme. Therefore, a large population still remains exposed to risk of HBV infection, and national and global eradication of HBV infection will remain a dream. Failure of seroprotection after receiving the HBV vaccination in the EPI will also be responsible for projecting a sense of false protection among vaccine recipients.
Dosing schedule is an important factor in the development of an antibody response and titre levels. According to the Advisory Committee on Immunization Practices (ACIP) of America, there should be a minimum gap of 8 weeks between the second and third doses and at least 16 weeks between the first and third doses of the HB vaccination23. To minimize frequent visits and improve compliance, the dosing schedule has been negotiated in the EPI to 6, 10 and 14 weeks24. Although some studies have shown this schedule to be effective, the GMT of anti-HBs antibodies achieved was lower than that achieved by the standard WHO schedule25. This may be one explanation of lower rates of seroprotection in our study. The GMT achieved in our study among the children having protective levels of antibodies is 85.81 IU/L which is lower than most other studies. This supports the observation that GMT achieved in this schedule is lower than that produced by the standard WHO schedule. This may result in breakthrough infection of HB in vaccinated individuals in later life due to waning immunity. However, the immune memory hypothesis supports protection of vaccinated individuals in later life in spite of low anti-HBs antibody titres26. Yet further studies are required to dispel this risk.
Another shortcoming of this schedule is to miss the dose at birth (‘0 dose’). It has been reported that the 0 dose of the HB vaccine alone is 70% - 95% effective as post-exposure prophylaxis in preventing perinatal HBV transmission without giving HB immunoglobulins27. This may also be a factor contributing to lower rates of seroprotection in our study as we have not done HBsAg and other relevant tests to rule out HBV infection in these children. Moreover pregnant ladies by and large are not screened for HBV infection in Pakistan routinely in the public sector except in a few big cities like Islamabad, Lahore or Krachi. Therefore, we do not know the HB status of pregnant mothers and the risk of transmission to babies remains high. Different studies have reported much varied figures of HB status in pregnant ladies. A study from Karachi reports 1.57% pregnant ladies are positive for HBsAg while a study from Rahim Yar Khan reports this figure to be up to 20%28,29. A study by Waheed et al regarding the transmission of HBV infection from mother to infants reports the risk to be up to 90%30. All of these studies support the importance of the birth dose of the HB vaccination and augment the fact that control and eradication of HB with the present EPI schedule is not possible. Jain from India has reported a study using an alternative schedule of 0, 6 weeks and 9 months. He has reported it to be comparable to the standard WHO schedule of 0, 1, 6 months in regards to seroprotection and GMT levels achieved31. This schedule can be synchronised with the EPI schedule, avoiding extra visits and incorporating the birth dose. A similar schedule can also be incorporated in our national EPI.
In our study, seroprotection rates were found to be low in the female gender and the difference was significant. This finding differs with other studies which report lower seroprotection rates in males32. Although the number of female children was less, there is no plausible explanation for this observation. The site of inoculation of the HB vaccine is also very important for an adequate immune response. Vaccines given in the buttocks or intradermally produce lower antibody titres than intramuscular injections given in the outer aspect of the thigh in children, due to poor distribution and absorption of the vaccine within the host body. The practice of giving vaccinations in the buttocks by vaccinators is a common observation which they feel convenient for intramuscular injection in children. This may also be one reason for low seroprotection rates in our study, as we picked the children at random who had received vaccination at public health facilities except a small number of private cases.
The effectiveness of the vaccine also depends on the source of procurement and proper maintenance of the cold chain. In this study 100% seroprotection was observed in children who received the HB vaccine procured from a private source. Although the number of private cases was less, this factor of source and the cold chain also needs attention. To address this issue proper training of EPI teams regarding maintenance of temperature, injection techniques, motivation and monitoring can improve outcomes substantially.
The findings of this study are different from published literature because this is a cross-sectional observational study. This reports the actual seroprotection rates after receiving the HB vaccination in the EPI schedule. While most other studies show the results after ensuring control of influencing factors such as type of vaccine, dose, schedule, route of administration, training and monitoring of local EPI teams and health status of vaccine recipients, etc. Therefore, this is an effort to look at a practical scenario and evaluate outcomes which can help in framing future guidelines to achieve the goal of control and eradication of HB infection. Further studies are required at a large scale to determine the effect of HB vaccination at a national level.
Conclusion
The HB vaccination programme has decreased the global burden of HBV infection, but evidence of decreased burden is not uniform amongst world population.Of course figures witness marked decrease in developed world while in developing world statistics show little change. Unfortunately, implementation of this programme is not uniformly effective in all countries, thus resvoirs of infection and the source of continued HBV transmission persists. HBV infection is moderately endemic in Pakistan. The HB vaccine has been included in the national EPI since 2004. The present study shows seroprotection rates of only 68.6% in vaccine recipients, which is low when compared with other studies; 31.4% of vaccine recipients remain unprotected even after vaccination. Moreover GMT achieved in seroprotected vaccine recipients is also low (85.81 IU/L). There can be multiple reasons for these results, such as type of vaccine used, maintenance of the cold chain, route and site of administration, training and monitoring of EPI teams and dosing schedule. In present practice, the very important birth dose is also missing. These observations warrant review of the situation and appropriate measures to be taken to rectify the above mentioned factors, so that desired seroprotection rates after HB vaccination in the EPI can be achieved among vaccine recipients.
The clinical features of early HAT are well defined, yet the features of delayed HAT are less clear. Delayed HAT is a rare complication of OLT that may present with biliary sepsis or remain asymptomatic. Sonography is extremely sensitive for the detection of HAT in symptomatic patients during the immediate postoperative period. However, the sensitivity of ultrasonography diminishes as the interval between transplantation and diagnosis of HAT increases due to collateral arterial flow. MRA is a useful adjunct in patients with indeterminate ultrasound exams and in those who have renal insufficiency or an allergy to iodinated contrast.
In the absence of hepatic failure, conservative treatment appears to be effective for patients with HAT but retransplantation may be necessary as a definitive treatment.
Case Presentation:
A 52 year old male with a history of whole graft OLT for primary sclerosing cholangitis presented with two days of fever, nausea, and mild abdominal discomfort.
One week prior to presentation, he was seen in the liver clinic for regular follow-up. At that time, he was totally asymptomatic and his laboratory workup including liver function tests were within normal range.
He has undergone OLT three years prior. At the time of transplant he required transfusion of 120 units of packed red blood cells, 60 units of fresh frozen plasma and 100 units of platelets due to extensive intraoperative bleeding secondary to chronic changes of pancreatitis and severe portal hypertension, but had an otherwise uneventful postoperative recovery.
On physical examination the temperature was 39C, heart rate was 125 beats per minute, respiratory rate was 22 bpm. Initial laboratory workup revealed a white blood cell count of 25,000/mm3, AST of 6230 U/L, ALT of 2450 U/L, total bilirubin of 11 mg/dL , BUN 55 mg/dL and Creatinine of 4.5 mg/dL. Lactate level was 5 mmol/L. Doppler ultrasonography revealed an extensive intrahepatic gas (Image 1A). Computed tomography of the abdomen and pelvis revealed extensive area of hepatic necrosis with abscess formation measuring 19x14 cm with extension of gas into the peripheral portal vein branches (Image 1B,C). Upon admission to the hospital, the patient required endotracheal intubation, mechanical ventilator support and aggressively fluid resuscitation. He was started on broad-spectrum antibiotics and a percutaneous drain was placed that drained dark, foul smelling fluid. Cultures from the blood and the drain grew Clostridium perfringens.
Magnetic resonance imaging (MRI), MRA revealed occlusion of the hepatic artery 2 cm from its origin and also evidence of collaterals (Image 2A,B).
Image 1: (Pannel A) Doppler ultrasonography reveal extensive intrahepatic gas. (Pannel B&C) Computed tomography of the abdomen and pelvis reveal an extensive area of hepatic necrosis with abscess formation measuring 19x14 cm with extension of gas into the peripheral portal vein branches.
Image 2: MRI & MRA reveal occlusion of the hepatic artery 2 cm from its origin and also evidence of collaterals.
Following drain placement, the patient’s clinical condition markedly improved with significant reduction of liver function test values. Retransplantation was considered but delayed in the setting of infection and significant clinical and laboratory testing improvement.
The patient was transferred to the medical floor in stable condition, and the drain was then removed.
A week later the patient developed low grade fevers and tachycardia. One day later he began to experience mild abdominal discomfort and high grade fevers. Repeat CT of the abdomen revealed worsening hepatic necrosis and formation of new abscesses. His clinical condition decompensated quickly thereafter requiring endotracheal intubation, mechanical ventilation and aggressive resuscitation. Percutaneous drain was placed and again, drained pus-like, foul-smelling material. His overall condition deteriorated, and he eventually expired a few days later.
Discussion:
Delayed (more than 4 weeks after transplantation) HAT is a rare complication of OLT with an estimated incidence of at around 2.8%1.
Risk factors associated with development of HAT include Roux-en-Y biliary reconstruction, cold ischaemia and operative time, the use of greater than 6 units of blood, the use of greater than 15 units of plasma, and the use of aortic conduits on arterial reconstruction during transplant surgery2.
Collateralization is more likely to develop after Live Donor Liver Transplantation (LDLT) than after whole-graft cadaveric OLT3. Therefore, the latter is also associated with increased risk of late HAT.
Although the clinical features of early HAT are well described, the features of delayed HAT are less clearly defined1: the patient may present with manifestations of biliary sepsis or may remain asymptomatic for years. Right upper quadrant pain has been reported to occur in both immediate and delayed HAT. The clinical presentations may include recurrent episodes of cholangitis, cholangitis with a stricture, cholangitis and intrahepatic abscesses, and bile leaks1. Doppler ultrasonography has been extremely sensitive for the detection of HAT in symptomatic patients during the immediate postoperative period but becomes less sensitive as the interval between transplantation and diagnosis of HAT increases because of collateral arterial flow4.
3D gadolinium-enhanced MRA provides excellent visualization of arterial and venous anatomy with a fairly high technical success rate. MRA is a useful adjunct in patients with indeterminate ultrasonography examination in patients who have renal insufficiency or who have allergy to iodinated contrast 5.
Antiplatelet prophylaxis can effectively reduce the incidence of late HAT after liver transplantation, particularly in those patients at risk for this complication6. Vivarelli et al reported an overall incidence of late HAT of 1.67%, with a median time of presentation of 500 days; late HAT was reported in 0.4% of patients who were maintained on antiplatelet prophylaxis compared to 2.2% in those who did not receive prophylaxis6. The option of performing thrombolysis remains controversial. Whether thrombolysis is a definitive therapy or mainly a necessary step in the proper diagnosis of the exact etiology of HAT depends mostly on the particular liver center and needs further analysis7. Definitive endoluminal success cannot be achieved without resolving associated and possible instigating underlying arterial anatomical defects. Reestablishing flow to the graft can unmask underlying lesions as well as assess surrounding vasculature thus providing anatomical information for a more elective, better plan and definitive surgical revision7. Whether surgical revascularization compared to retransplantation is a viable option or only a bridging measure to delay the second transplantation has been a longstanding controversy in the treatment of HAT.
Biliary or vascular reconstruction do not increase graft survival and ongoing severe sepsis at the time of re-graft results in poor survival7. However, although uncommon, delayed HAT is a major indication for re-transplantation7. In the absence of hepatic failure, conservative treatment appears to be effective for patients with hepatic artery thrombosis.
C. perfringensis an anaerobic, gram-positive rod frequently isolated from the biliary tree and gastrointestinal tract. Inoculation of Clostridium spores into necrotic tissue is associated with formation of hepatic abscess8.
Necrotizing infections of the transplanted liver are rare. There have been around 20 cases of gas gangrene or necrotizing infections of the liver reported in the literature. Around 60% of these infections were caused by clostridial species with C. perfringens accounting for most of them. Around 80% of patients infected with Clostridium died, frequently within hours of becoming ill9,10. Those who survived underwent prompt retransplantation and the infection had not resulted in shock or other systemic changes that significantly decreased the likelihood of successful retransplantation8.
Because the liver has contact with the gastrointestinal tract via the portal venous system, intestinal tract bacteria may enter the liver via translocation across the intestinal mucosa into the portal venous system. Clostridial species can also be found in the bile of healthy individuals undergoing cholecystectomy9,10.
The donor liver can also be the source of bacteria. Donors may have conditions that favor the growth of bacteria in bile or the translocation of bacteria into the portal venous blood. These conditions include trauma to the gastrointestinal tract, prolonged intensive care unit admissions, periods of hypotension, use of inotropic agents, and other conditions that increase the risk of potential infection 8,9,10. C. perfringens sepsis in OLT recipients has been uniformly fatal without emergent retransplantation. Survival from C. perfringens sepsis managed without exploratory laparotomy or emergency treatment has been extremely rarely reported8. In those patients who survived, and in whom the infection has not resulted in shock or multiple organ failure, retransplantation may be successful8.
Although our patient survived his intensive care course, his recovery was tenuous as he quickly developed additional hepatic abscesses that led to his eventual demise. Post-mortem examination in our patient revealed intra-hepatic presence of Clostridium perfringens.
He was managed conservatively since he markedly improved both clinically and by liver function tests. Because of this, retransplantation was delayed. He was also already on antiplatelet prophylaxis.
Conclusion:
We report an interesting case of Clostridium perfringens hepatic abscess due to late HAT following OLT. Although the patient initially improved with non-surgical treatment, he eventually died. In similar cases, besides aggressive work-up and medical management retransplantation may be necessary for a better long term outcome.
Many subjects with chronic Hepatitis C Virus (HCV) infection show persistently normal alanine aminotransferase (ALT) levels (PNALT),1-4 and thus formerly defined as ‘healthy’ or ‘asymptomatic’ HCV carriers.1 However, it is now clear that only a minority of these people show normal liver (15-20%).5-7 Therefore, ‘normal ALT’ does not always mean ‘healthy liver.’4
It is known that during the course of HCV infection ALT levels could fluctuate widely, with long periods of biochemical remission.1-4Thus, at least two different subsets of HCV-PNALT carriers exist: patients with temporal ALT fluctuations, that could be within the normal range for several months, and true ‘biochemically silent’ carriers showing persistently normal ALT values.4It means that the observation period should not be shorter than 12 - 18 months, and ALT determinations should be performed every 2 - 3 months.4, 6
Although liver damage is usually mild, 1, 2the presence of more severe chronic hepatitis (CH) or cirrhosis has been reported despite consistently normal liver biochemistry.8Although some studies showed that HCV carriers with normal ALT have mild and rather stable disease, others reported a significant progression of fibrosis in approximately 20-30% of the patients with ALT normality.9The development of hepatocellular carcinoma (HCC) has been also described.10Sudden worsening of disease with ALT increase and histological deterioration has been reported after many years of follow-up.11
Finally, HCV carriers with PNALT may suffer from extra-hepatic manifestations, sometimes more severe than the underlying liver disease: lymphoproliferative disorders, mixed cryoglobulinaemia, thyroid disorders, sicca syndrome, porphyria cutanea tarda, lichen planus, diabetes, chronic polyarthritis, etc.1, 2, 12
Therefore, the possibility of progression to more severe liver damage despite persistently normal biochemistry, the risk of HCC, the possibility of extra-hepatic diseases, and economic considerations, suggest that HCV-infected persons with PNALT should not be excluded a priori from antiviral treatment.1, 2
The earliest guidelines discouraged interferon (IFN) treatment in patients with PNALT because of the cost and side effects of therapy,1, 2 and of the low response rates to IFN monotherapy (<10-15%) with a risk of ALT flares in up to 50% of patients during treatment.9
The introduction of the combination of weekly subcutaneous pegylated-IFN (PEG-IFN) plus daily oral ribavirin (RBV) has led to response rates >50%, with a favourable risk-benefit ratio even in patients with slowly progressing disease.1, 2, 9 The first trial of PEG-IFN plus RBV found a sustained virological response (SVR) in 40% of HCV-1 carriers with PNALT treated for 48 weeks, and in 72% of HCV-2 and HCV-3 treated for 24 weeks.13The efficacy of antiviral treatment with PEG-IFN plus RBVwas subsequently confirmed in clinical practice.14, 15
However, in everyday practice, management of carriers with PNALT may be paradoxically more difficult than that of patients with abnormal ALT levels. Indeed, it is not always so easy to ascertain in the single case whether it should be considered as healthy subject or true patient. Several topics to date remain unresolved: Should these ‘seemingly healthy’ people undergo routine liver biopsy? Is antiviral treatment justified in ‘asymptomatic’ subjects with persistently normal liver biochemistry? Is long-term follow-up needed in this setting, and how long it should last?2
Liver biopsy provides helpful information on liver damage, as it may reveal the presence of advanced fibrosis or cirrhosis. Without a biopsy, it is impossible to clinically distinguish true ‘healthy’ carriers from those with CH.4 On the other hand, it is difficult to recommend routine biopsy for all HCV-PNALT .4 The decision to perform a biopsy should be based on whether treatment is being considered, taking into account the estimated duration of infection, probability of disease progression, willingness to undergo a biopsy, motivation to be treated, and availability of non-invasive tools to assess liver fibrosis.12 The recently developed transient elastography has improved our ability to non-invasively define the extent of fibrosis in HCV persons.5
Careful evaluation of parameters associated with disease progression is mandatory to assess the actual need for antiviral treatment.4 Indeed, it is really impossible to suggest antiviral therapy in all HCV carriers, as the costs would be exceedingly high, due to the high number of HCV patients with PNALT. Data from the literature indicate that the main factors of progression are male gender, advanced age, severe fibrosis, ALT flares, and steatosis.1-2
Cost/benefit might be particularly favourable in:
Young patients, having high rate of SVR (e.g. females, low viral load, HCV genotype non-1, etc).
Middle age patients with ‘significant’ liver disease and/or co-factors of progression of liver damage, thus at risk of developing more severe liver disease.12
The age issue has a critical role for decision making. Younger patients have a higher chance of achieving SVR and tolerating therapy better; they a have longer life expectancy, are often well motivated, usually have minimal disease and fewer contraindications. Thus, in this group decision to treat should be based more on expected response and motivation than on the severity of liver disease.
On the contrary, older patients respond less well to therapy, are more likely to have significant liver disease and/or co-factors, could experience more side effects and may be less motivated. Thus, in this group decision to treat should be based on the severity of liver disease and on the possibility of SVR.
A recent Italian Expert Opinion Meeting suggested the following recommendations:12
HCV carriers with PNALT may receive antiviral treatment with PEG-IFN plus RBV using the same algorithms recommended for HCV patients with abnormal ALT.
Decision making should rely on individual characteristics such as HCV genotype, histology, age, potential disease progression, probability of eradication, patient motivation, desire for pregnancy, co-morbidities, co-factors, etc.
Treatment might be offered without liver biopsy in patients with a high likelihood of SVR (e.g. age <50 years + non-1 HCV genotype + low viral load), in the absence of co-factors of poor responsiveness.
Inpatients aged 50–65 years, and in those with a reduced likelihood of achieving a response, biopsy may be used to evaluate the need for therapy, with treatment being recommended only for patients with more severe fibrosis and higher possibility of SVR. Biopsy and therapy are not recommended in the elderly (>65-70 years).
In patients who are not candidates for antiviral treatment, follow-up may be continued, and ALT should be monitored every 4-6 months. Avoidance of alcohol and obesity may be strongly recommended.12 It is not clear whether these subjects should be routinely offered anti-HBV vaccine, given the risk of disease progression in the case of HBV infection.12 Antiviral treatment should be re-considered in the case of ALT flares, US abnormalities or platelet count decrease. Repeated measurements of serum HCV RNA to evaluate disease progression is not recommended.1, 9, 11, 12.
Hyperthyroidism is one of the most frequently encountered conditions in clinical endocrinology.1 The modes of treatment available are antithyroid drugs, surgery and radioiodine (RAI) and although each of these is highly successful in controlling or curing hyperthyroidism none leads to permanent euthyroidism on a consistent basis. 2 Although over the last three decades RAI therapy has replaced surgery as the leading form of definitive treatment 3,4, 5 there is no universally accepted dose or regime for its use. Previous attempts to individualise the dose of RAI to reduce the rate of post-RAI hyper- or hypothyroidism have been unsuccessful 6, 7. Fixed dose RAI administration has therefore become the most commonly used regime although the actual dose of RAI used varies considerably and ranges between 185MBq to 600MBq 8, 9. For the last two decades we have used a fixed RAI dose of 550MBq for all patients. Others have used this regime with a high success rate 10 and a prospective head to head comparison with the calculated dose method found the fixed dose regimen to be superior for curing Graves’ hyperthyroidism 11.
Conflicting results have been produced in several studies that have attempted to predict outcome following RAI therapy by correlating cure rate with various pre-treatment factors including age, gender, aetiology of hyperthyroidism, goitre size, use of antithyroid drugs, free thyroxine levels at diagnosis and thyroid antibody status. Various forms of calculated or low fixed dose RAI therapy have been used in these studies but no study used a high fixed dose of 550MBq. In this study we have evaluated the overall success rate of high fixed dose RAI therapy and attempted to identify simple clinical predictors of failure to respond the initial RAI dose.
Patients and Methods
The study is a retrospective analysis of 584 consecutive patients referred to the Shropshire endocrinology service (Princess Royal Hospital and Royal Shrewsbury Hospital) over a 14 year period for the treatment of hyperthyroidism. These patients received RAI therapy at Royal Shrewsbury Hospital, which is the only centre providing facilities for RAI administration in the county of Shropshire and also draws referral from adjoining trusts in Powys, North Wales. Information for this study was obtained from the thyroid database which is maintained on all patients who have received RAI since 1985 at the above hospitals.
RAI was administered both as a primary (53%) and as secondary (47%) treatment. A majority of patients with moderate to severe hyperthyroidism were rendered euthyroid by antithyroid drugs (ATD). Ninety percent (518/584) patients were pre-treated to euthyroidism by antithyroid drugs (carbimazole in 95% and propylthiouracil in 5%) before RAI therapy. Carbimazole was withdrawn one week and propylthiouracil 4 weeks prior to RAI therapy. A standard RAI dose of 550MBq was administered to all patients without a prior uptake study. Thyroid function was measured at 6 weeks and at 3, 6 and 12 months following RAI therapy. ATD drugs were not recommenced routinely following RAI therapy and were reserved for patients who were persistently and significantly hyperthyroid following RAI administration. Patients who developed clinical and biochemical hypothyroidism after the initial 6-8 weeks were commenced on thyroxine. Patients with high free thyroxine level (FT4) and a suppressed thyroid stimulating hormone (TSH) level and those on antithyroid medication were defined as being hyperthyroid, those with low FT4 or on thyroxine as hypothyroid and those with normal FT4 and a normal or low TSH as euthyroid. At the end of one year if a patient remained hyperthyroid, another RAI dose of 550MBq was administered. The patient was considered to have been “cured” if euthyroidism or hypothyroidism was achieved during the first year following RAI therapy and “not cured” if patient remained persistent hyperthyroidism at the end of this period.
Information recorded on the database included age, gender, aetiology, indication (primary or secondary), dose of RAI, number of RAI doses, name and duration of antithyroid drugs used, if any, and FT4 and TSH levels at diagnosis, at the time of RAI therapy and at 6 weeks, 3, 6 and 12 months after RAI therapy. Diagnosis of Graves’ disease was based on the presence of Graves’ ophthalmopathy or a combination of a diffuse goitre and a significant titre of thyroid peroxidase antibodies or if radionuclide scan showed diffuse uptake. Toxic nodular disease was diagnosed on the grounds of a nodular goitre and a focal increase in radionuclide uptake. Patients who could not be classified to either of the groups on clinical grounds and where a radionuclide scan could not be performed for a variety of reasons, were categorised as “unclassified” on aetiological grounds.
Statistical analysis
Continuous random variables were compared using t-tests and association of categorical variables by using chi-squared tests. The effect on outcome (cure of hyperthyroidism) of all variables was assessed by using logistic regression analysis and a step-wise routine was applied to choose the best set of predictors. All analyses were carried out by using NCSS2000.
Results
Data on 584 patients was included with a mean age of 56 years (range 20-90) and a female preponderance (82%). Assessment of the aetiology of hyperthyroidism was made by the above-mentioned criteria. In 110(15%) patients precise aetiological diagnosis could not be made. 344/474 (72%) patients had hyperthyroidism secondary to Graves’ disease and 134/474(28%) had toxic nodular disease. 518 patients received pre-RAI antithyroid medications. Mean free thyroxine level at time of diagnosis was 45.4pmol/L in 259 patients in whom this information was available. Data for thyroid status at 3, 6, and 12 months post-radioiodine were available in 97, 94 and 100% patients respectively (see Table 1).
Table 1: Thyroid status at 3, 6 and 12 months
Euthyroid (%)
Hypothyroid (%)
Hyperthyroid (%)
3 months
308 (54%)
176 (31%)
87 (15%)
6 months
210 (38%)
280 (51%)
59 (11%)
12 months
134 (23%)
411 (70%)
39 (7%)
FT4 values were entered onto the database more recently and this result was available in 259 patients. The group of patients where FT4 data was available was comparable to the group where this information was not available in all respects apart from age (mean age (SD) 54 (±15) vs 58 (±14) years respectively, p<0.02). Similarly, the group of patients in whom the aetiology could not be ascertained was not different from the group where the aetiology could be identified in any respect apart from the age (mean age (SD) 60 (±13) vs 55 (±15) respectively).
Table 2 – Forward Stepwise (Wald) logistic regression analysis to identify factors independently associated with failure to respond to first dose of RAI
Variables
P value
Adjusted r2; OR (95% CI)
Free T4 at diagnosis
0.005
0.084; 1.04 (1.01-1.07)
Free T4 > 45 pmol/l at diagnosis*
0.02
0.056; 3.43 (1.17-10.04)
Age
0.81
N/A
Gender
0.18
N/A
Aetiology
0.23
N/A
Pre RAI use of anti-thyroid drugs
0.42
N/A
* Regression analysis carried out with free T4 as a continuous variable and separately as a categorical variable at a cut off of 45pmol/l
One year following RAI treatment, 543(93%) patients were either euthyroid (162;28%) or hypothyroid (383;65%) and considered “cured”; 39(7%) patients remained hyperthyroid and required further doses of RAI, with 34(6%) patients requiring two doses and 5(1%) patients three doses. At 3 months, 484 out of 571 (85%) patients, and at 6 months, 490 out of 549 (89%) patients were “cured” (table 2). On univariate analysis no correlation could be established between the failure to respond to the first dose RAI and age, gender, aetiology or use of antithyroid medication (p = ns for all) although the rate of hypothyroidism was significantly higher at the end of one year in patients with Graves’ disease as compared to those with toxic nodular disease (77.1% vs. 50.3%, p<0.01). These results were not affected by limiting the analyses to any of the following groups: only those patients in whom the aetiological diagnosis could be made (n=478), only those patients in whom FT4 value was available (n=259) or only those patients where both FT4 was available and aetiology could be ascertained (n=209). On univariate analysis FT4 at diagnosis was associated with the outcome when it was used as a continuous variable (p<0.05) or as a categorical variable with the cut off set at mean FT4 value of 45pmol/L (p=0.01) and high values were associated with failure to respond to the first dose of RAI (mean ± SD, 57.28±20.1 v 44.58±16.1 pmol/L, p<0.05). On multivariate analysis with all variables, FT4 was found to be independently associated with outcome and again this association was seen when FT4 was used as a continuous variable (p=0.01) as well as a categorical variable (p=0.02). On using step-wise selection routine only FT4 could be chosen as a predictor when criterion for selection was set at p=0.05 and a value of over 45pmol/L predicted failure to respond to the first dose of RAI.
Discussion
The use of a standard fixed-dose RAI therapy is gaining increasing popularity and several studies have now shown that formal estimation of the required dose based on the thyroid size and iodine kinetics does not lead to a higher cure rate 6,7,10,11 or a lowerhypothyroidism rate 7. For several years we have used 550MBq dose for all patients of hyperthyroidism. The overall success rate with this regime was 93% and only 7% of patients required a repeat RAI dose. These figures are comparable to those from most other centres, which have used a similar dose of RAI 10. In addition to achieving a high cure rate, hyperthyroidism was controlled rapidly with 85% of the patients becoming either euthyroid or hypothyroid within 3 months of treatment. Early onset of hypothyroidism (>70% at 12 months) facilitated institution of thyroxine replacement therapy during the first year during which the patients were being closely followed.
The use of a relatively higher dose of RAI leads to more stringent restrictions to the normal life of patients and these have to be followed for a longer period of time than is the case with the use of a lower dose. Majority of patients accept these restrictions at the prospect of a cure of hyperthyroidism. However, even at this dose, 7% of patients required repeat dosing which in turn led to another restrictive period for these patients. In view of this it is useful to be able to predict failure of the first dose in an individual patient. This would enable us to warn these patients about the higher possibility of requiring repeat dosing, further period of post-RAI restrictions and target them for a closer follow up. To allow us to make this prediction we correlated simple clinical pre-treatment variables to the need for repeat dosing. We found that there was no statistically significant correlation between age, gender, aetiology and the use of anti-thyroid medication prior to RAI and the outcome following RAI therapy although a high free thyroxine level at diagnosis predicted a failure of the first dose to achieve a cure of hyperthyroidism. There are several conflicting reports in the literature on the correlation between these factors and the response to RAI therapy. Most of the studies have failed to show a significant association between the age of the patient and the outcome irrespective of whether the age was used as a continuous or a categorised variable 12-15 although in a study where a standard 150 gray RAI was used age >50 was found to be associated with a higher failure rate 16. In one study, male gender was associated with a lower cure rate following a single dose of RAI in patients with Graves’ disease 12 although others have failed to confirm this association 13,14. Use of antithyroid drugs prior to RAI has been shown to independently reduce the success rate of RAI 17, 18 while other studies have shown such an association with the use of propylthiouracil but not with carbimazole 19, 20. Literature on the association between the aetiology of hyperthyroidism and the outcome is even more confusing. Patients with toxic nodular disease have been considered to be more radio-resistant as compared to patients with Graves’ disease 21 although opposite results have also been noted 22. In other studies no correlation could be established on multivariate analysis between the aetiology and outcome following RAI 14, 18. Our study is the only one which analyses the influence of these factors on the outcome following the use of a standard 550MBq RAI dose and the above studies which have attempted to identify clinical predictors of outcome have either used various forms of the calculated dose regime or a lower fixed-dose RAI regime. We feel that this is the reason for the inconsistencies in the results and when a 550MBq dose RAI is used only FT4 value at diagnosis could predict the failure of RAI therapy to achieve cure. This dose of RAI appears to override the variations in the response induced by the remaining pre-treatment variables studied.
Studies using smaller doses or calculated doses of RAI have shown the outcome to be inversely associated with the thyroid size 14, 16 although this could not be ascertained in our study due to the lack of consistent documentation of the size ofgoitre in the clinical notes. In addition there are several possible confounding factors. Firstly the overall cure rate could have been influenced by the long period of time over which patients have been included (15 years) and the resulting changes in the criteria and threshold for the use of RAI. However if we divide the figures into 3 time periods of 5 years each, the findings remain consistent during each of these periods. Secondly, in over 50% of our patients, RAI was administered as a primary measure and it could be argued that a larger number of patients with milder hyperthyroidism may have been included in our cohort as compared to the patients at other centres where RAI is mainly reserved for patients who fail to respond to ATD. However there was no significant difference in the cure rate between those patients who received RAI as a primary measure and those in whom RAI was administered as a secondary treatment (94% v 93%). Thirdly in 15% of patients the aetiology could not be ascertained by using our well-defined criteria, mainly because of the practical difficulty of performing radionuclide scans in some of the patients where the diagnosis could not be made clinically. We do not feel that our results on the association between the aetiology and the cure rate were affected, as the patients with undefined aetiology were comparable to the remaining patients in all respects apart from age and had similar outcomes. Lastly the information on the FT4 value at diagnosis was available in only 259 patients. To exclude a selection bias this group was compared to the group of patients where this information was not available. Again the only difference between the two groups was the age distribution. In both instances this difference was not large (though statistically significant) and we do not feel it affected the outcome, especially as age does not appear to influence the outcome following RAI therapy. We could not assess the impact of post-RAI use of antithyroid drugs as these were not routinely restarted following RAI therapy at our centre.
In conclusion, high fixed dose RAI therapy is a very effective treatment for patients with hyperthyroidism and has a high success rate. Failure to respond to this dose cannot be predicted by most of the pre-treatment variables apart from the severity of the hyperthyroidism as judged by the FT4 value at diagnosis. Patients who present with severe hyperthyroidism should be warned regarding the higher possibility of requiring further doses of radioiodine even when treated with a dose of 550MBq.
First described in 1971 by Spillane et al.1, painful legs and moving toes (PLMT) is a syndrome consisting of pain in lower legs with involuntary movements of the toes or feet. Pain varies from moderate discomfort to diffuse and deep and usually precedes movements by days to years. The movements themselves are often irregular and range from flexion/extension, abduction/adduction to clawing/straightening and fanning/ circular movements of the toes.1,2 This syndrome may affect one leg or spread to involve both legs.3
PMLT incidence and prevalence remain largely unknown since it is still a relatively rare disorder worldwide. Age of onset is between the second and seventh decades of life. It has been postulated that lesions of the peripheral or central nervous system after nerve or tissue damage might lead to impulse generation that subsequently causes the symptoms seen in PLMT.4 We report a case of PMLT that presented to our Neurology Movement Disorder Clinic along with a discussion on the pathophysiology, differential diagnosis and clinical management of this rare debilitating condition.
Case report
63 year old, morbidly obese (BMI 41.7) Caucasian male patient with past medical history of stroke 10 years ago, on long term anticoagulation, hypertension, type II controlled diabetes mellitus, asbestos exposure, bilateral hip and knee osteoarthritis, left total knee replacement 2 years ago, and non-traumatic ruptured Achilles tendon; presented with complaints of involuntary movements in both legs over the last 8-10 years. He had unprovoked flexion and extension of the toes along with feet movement at all times with no diurnal variation. He admitted to having a constant severe pain described as 'twisting a rubber band' with 10/10 intensity that radiated up to his calf accompanied by numbness and dorsal swelling of both feet for many months. He claimed to have partial relief whilst walking but had difficulty walking without a cane as he ‘“could not balance with constantly moving [his] feet”’. Tylenol 500mg as required and amitriptyline 20mg at night prescribed by his primary care physician provided no relief.
He also has a history of snoring, daytime fatigue, and non-restorative sleep with frequent nocturnal awakenings due to bilateral feet pain. He recalled having a stroke with transient confusion and focal hand weakness along with visual problems about 10 years previously. All laboratory and radiological investigations were negative and he recovered fully. He had previously served with the US armed forces and had been exposed to ‘Agent Orange’ in Vietnam.
He had no medical allergies and his current medications include amitriptyline 25mg at night, hydrochlorothiazide 25mg once daily, lisinopril 10mg once daily, loradatine 10mg once daily, metoprolol tartrate 20mg twice daily, simvastatin 20mg once daily, vitamin B complex one tablet once daily and warfarin once daily. He denied any history of alcohol, tobacco, or recreational drug abuse in the past. His mother had a history of hypertension and chronic low back pain; no members of his family had any neurological or movement disorders.
Physical examination revealed an alert, awake, and well oriented male with bilateral lower extremity varicose veins. He was observed to have semi-rhythmic flexion-extension and occasionally abduction movements of the phalanges, especially in the great toes. There was a profound decrease in vibration sense below both knees and it was almost absent on both feet, decreased reflexes in both feet, and absent proprioception in the phalangeal joints. He was also observed to have decreased pinprick and monofilament sensation in both legs below the knee. Bilateral ankle reflexes were diminished with negative Babinski sign. Both lower extremity dorsalis pedis and posterior tibial pulsations were palpable. He did not have any cerebellar signs. He did have pitting oedema up to his shins in both lower extremities, extending from his feet to upper one third of the legs. There were no abnormalities noted on the bilateral lower extremity EMG and there was no electrodiagnostic evidence of large-fiber neuropathy.
He was diagnosed with painful legs and moving toes syndrome and started on a trial of gabapentin 300mg at night. He was advised to increase it to 1200mg and to continue taking his amitriptyline 25mg at night. Scheduled MRI of the brain could not be done due to his morbid obesity. He was arranged follow up in three months in the clinic.
Methods
A review of published literature on PLMT was done using MEDLINE and PubMed databases. Searches were conducted to find articles from 1971 – 2010. Medical subject headings used to search the databases included PLMT with subheadings of Movement disorder, Electromyography, and Polysomnography as well as keyword search using ‘PLMT’. Single author reviewed titles and abstracts of potentially relevant articles.
Review of current literature
We reviewed approximately 19 PLMT articles that have been published to date with a total of 72 patients: 30.5% males, 69.5% females (median age 55 & 64 years,
respectively). Clinical presentations in the majority of the cases were burning pain in lower extremities and involuntary movements of the toes. The most common predisposing conditions were neuropathy and radiculopathy (see Table 1).
Table 1 - Painful Legs & Moving Toes Syndrome ~ Review of Literature (1971- 2010)
Author
Year
Sex/ Subjects
Subject age
# of cases
Clinical presentation
Spillane et al
1971
M (4) F (2)
51, 52, 52, 53 66, 68
6
Burning/throbbing LE pain followed by writhing/clawing and flexion/extension movements of the toes
Dressler et al
1994
M (4) F (16)
28, 36, 54, 73 28-76
20
Pain in LE followed by involuntary flexion/extension and abducion/adduction of the toes
Shime et al
1998
F (1)
63
1
Involuntary flexion/extension of the toes bilaterally and aching/crampy pain in both feet
Schott et al
1981
M (1) F (4)
66 56, 57, 69, 77
5
Crushing pain in both feet followed by involuntary writhing and flexion/extension of the toes; burning pain in foot followed by writhing toe movements
Montagna et al
1983
M (1) F (2)
57 74, 76
3
Burning pain in one or both LE followed by involuntary flexion/extension, abduction/adduction, and fanning/clawing of the toes
Shime et al
1998
F (1)
63
1
Involuntary flexion/extension of the toes bilaterally and aching/crampy pain in both feet
Villarejo et al
2004
M (1)
66
1
Paresthesias/burning pain in both feet followed by involuntary flexion/extension and abduction/adduction of the toes
Aizawa et al
2007
F (1)
73
1
Tingling pain in both feet followed by involuntary abduction/adduction of the toes
Guimaraes et al
2007
M (1)
60
1
Wringing-like pain in in L foot and R leg followed by flexion/extension and abduction/adduction of the toes
Eisa et al
2008
M (1) F (1)
62 76
2
Burning pain in bilateral LE followed by semirhythmic flexsion/extension of the toes
Alvarez et al
2008
M (6) F (8)
25-84 (mean 69)
14
Burning pain of LE followed by involuntary flexion/extension, abduction/adduction, fanning, or clawing of the toes
Tan et al
1996
F (1)
57
1
Severe burning pain in both LE followed by involuntary flexion/extension and abduction of the toes
Dressler et al
1994
M (4) F (16)
28, 36, 54, 73 28-76
20
Pain in LE followed by involuntary flexion/extension and abducion/adduction of the toes
Yoon et al
2001
F (1)
56
1
Burning pain in R foot with flexion and lateral deviation of the toes
Miyakawa et al
2010
M (1) F (1)
36 26
2
Burning pain in R arm followed by involuntary flexion/extension of R thumb; pain in L leg accompanied by flexion/extension and abduction/adduction of L toes
Schoenen et al
1984
M (2) F (4)
49, 74 68, 69, 71, 80
6
Burning/aching pain in LE followed by involuntary flexion/extension and writhing of the toes
Sanders et al
1999
F (1)
76
1
Deep/throbbing pain in L leg followed by invloluntary flexion/extension and abduction/adduction of L toes
Ikeda et al
2004
F (1)
75
1
Involuntary flexion/extension of the toes bilaterally followed by pain in both legs
Kwon et al
2008
F (1)
75
1
Painless wriggling movements of the toes in both feet
Total Number of articles reviewed = 19 Total Number of Cases: Male = 22 (Median Age = 55 years); Female = 50 (Median Age = 64 years) Author/Article References in chronological order (Top to below): 1, 2, 3, 4, 5, 6, 7, 8, 9, 11, 16, 17, 23, 24, 25, 29, 31, 32, 33
In 1981 Schott GD et al reported that in 3 PLMT patients the EMG revealed evidence of denervation in the affected muscles. Montagna et al of the University of Bologna, Italy reported 3 cases of PLMT that exhibited evidence of peripheral neuropathy on EMG. Polysomnography (PSG) studies on these patients showed reduced movements during sleep with increase in slow wave or rapid eye movement sleep.5 This suggested the movements could have arisen centrally.
Guimaraes et al of the Universidade Nova Lisboa, Portugal reported one patient with a history of Hashimoto’s disease whose lower extremity EMG showed spontaneous arrhythmic bursts of the affected muscles during wakefulness which disappeared during sleep6. Both suggested the movements could have arisen centrally.
Alvarez et al of the Mayo Clinic described 14 cases of PLMT in 2008 in which burning pain often preceded the movements. PSG studies confirmed these movements would also persist in light stages of sleep which pointed to a central origin.7 Eisa et al of Yale University School of Medicine, Connecticut described 2 cases of PLMT in which one patient had a past history of lumbosacral root injury and the other systemic lupus erythematosus with peripheral neuropathy on EMG.8 Interestingly, in the latter patient her pain occurred years after the onset of involuntary toe movements.8
Discussion
Spinal cord and cauda equina diseases, neuropathies, radiculopathies, drugs and other systemic diseases are the main cause of this syndrome although many cases are still idiopathic. The most common predisposing conditions were neuropathy (i.e. polyneuropathy from alcoholism, hypertrophic mononeuritis, or tarsal tunnel syndrome) and radiculopathy.7 Other etiologies include nerve root lesions, peripheral nerve trauma, spinal ganglia lesions, cauda equina lesion, Wilson’s disease, herpes zoster myelitis, HIV, neuroleptics, and chemotherapeutic agents.9-19
The involuntary movements appeared bilaterally in the toes in our patient, which suggests that central reorganization (especially in the spinal level) is the cause of PLMT. EMG and nerve conduction studies have proven helpful in demonstrating spontaneous arrhythmic bursts of affected muscles and underlying neuropathy in some patients. Although the exact mechanism remains elusive, it has been proposed that impulses generated in lesioned peripheral nerve, posterior nerve root/ganglion, or afferent fibers pass into the spinal cord - some to higher areas to cause pain, while others into the local interneuron and motor neurons to generate involuntary movements of the toes.5
In patients with clinical or electrophysiological evidence of peripheral nerve or root problem, these lesions can initiate or even alter afferent input to the spinal cord and cause subsequent central and efferent motor reorganization, which may explain the limited
success these patients had with nerve blocks or lumbar sympathetic blockade.2 Similarly, some have suggested that even though the radiation of pain following local trauma seemed to resemble causalgia,20 there was a lack of hyperpathia and changes in the soft tissue, bones, and blood vessels as well as a poor response to sympathetic blockade, thus making clinical features of PLMT inconsistent with known radicular disorders.3
Interestingly, some believed that the central nervous system played an essential role in PLMT via a central oscillator.21 It has also been proposed that hyper-excitability of the damaged peripheral nerves could cause symptoms of PLMT by way of the sympathetic nervous system. More specifically, the sympathetic nervous system could potentially serve as a bridge between injured afferent fibers and sympathetic nerve fibers,22 allowing abnormal afferent impulse to travel to efferent fibers and ultimately leading to continuous pain with involuntary movements. This was evident in the fact that lumbar sympathetic ganglion blockade provided moderate symptomatic relief for some patients even though it was short-lived.4 Interestingly, one of the explanations put forth was the possibility of spinal/supraspinal reorganization,23 which coincided with the hypothesis of central reorganization mentioned above.
Clinical Management
Numerous treatments including antiepileptics, benzodiazepines, antispasmodic agents, and antidepressants have been tried with little success.1,2,24,25 However, temporary success was observed with local anesthetic nerve blocks, epidural blocks, sympathectomy/sympathetic blockade, neurectomies, botulinum toxin type A injection, transcutaneous electrical nerve stimulation, vibratory stimulation, and epidural spinal cord stimulation.1,2,15,26,27 Analgesics, steroids, anti-inflammatory agents, vitamin B12 injections, propranolol, quinine sulphate, and local anesthetics only offered temporary relief as well.3 GABAergic agents such as gabapentin and pregabalin were the most effective in attenuating the pain and the movements, possibly via both central and peripheral mechanisms.7,24,25 It has been reported that gabapentin as high as 600mg three times daily could control symptoms of PLMT long-term.25
Treatment of PLMT has also been attempted with botulinum toxin A at the level of lumbosacral roots and peripheral nerves with moderate relief of symptoms, although toe movements did return after a few months.8 It was suggested that botulinum toxin A might have acted via reduction of muscle spindle discharge leading to decreased central sensitization, as well as antisympathetic, antiglutamergic, or anti-inflammatory effects.28
Differential Diagnosis
The syndrome of PLMT exhibits certain features similar to the restless leg syndrome (RLS). In RLS the sensation in the legs could be burning, creeping, or tingling coupled with an urge to move them, especially early in the night. Movements such as walking or stretching relieve the symptoms whereas rest makes them worse. However, in PLMT pain is severe, constant, unrelated to the sleep-wake cycle, and is not relieved by
movements or walking.23 In addition, its involuntary movements of the toes or feet also differ from the myoclonic jerks of RLS.
In conditions such as thalamic syndrome and limb pain with myoclonus, patients may experience pain and involuntary movements as well but they often occur simultaneously as opposed to in PLMT where pain often precedes the movements.17 In disorders such as Parkinson’s disease and dystonia, sustained involuntary movements in the feet can be present and pain can be an associated feature. But the movements are typically sustained muscle contractions, which are different from the typical movements associated with PLMT.
Prognosis
PLMT is a newly discovered syndrome and since there has not been a systematic study following these patients long-term, it is currently quite difficult to predict the outcome of this syndrome and its effect on lifespan, though there has yet been a report of a patient actually dying from this syndrome. However, it is known that PLMT is a debilitating condition that greatly reduces patients’ quality of life.
Conclusion
Since Spillane et al first described it in 1971, there have been more reported cases of PLMT and its variants over the years. Though much progress has been made in elucidating its etiology, its exact mechanism still remains a mystery. Similarly, even though EMG and nerve conduction studies have proven helpful in demonstrating spontaneous arrhythmic bursts of affected muscles and underlying neuropathy in some patients, diagnosis of PLMT remains largely on history and clinical presentation.
Physicians should be aware of this rare debilitating condition. It is important to consider PLMT in a patient with painful legs and/or restless leg syndrome without any significant history of neurological disease or trauma. Treatments such as different combinations of medications and invasive techniques are complex and generally lead to a poor outcome.
Thyrotoxic periodic paralysis (TPP) is an uncommon disorder characterised by simultaneous thyrotoxicosis, hypokalaemia, and paralysis that occurs primarily in males of South Asian descent.1 Many affected patients do not have obvious symptoms and signs of hyperthyroidism and hence may be misdiagnosed or overlooked on presentation.2 We hereby report a male patient who presented to us with weakness of all four limbs. The patient was evaluated and diagnosed to be having TPP.
Case History
A 30-year-old male patient, who was an agriculturist by profession, presented with weakness of all four limbs of one-day duration. The weakness first appeared in his lower limbs and then in the upper limbs. There were no sensory symptoms or bladder involvement. He was not a known hypertensive, diabetic or thyrotoxic patient. He was not on any medication for any significant illness.
On general physical examination, there was no pallor, icterus, cyanosis, clubbing, lymphadenopathy or pedal oedema. Multinodular goitre was noted on thyroid examination. There was no exopthalmos, lid lag, pretibial myxoedema or other signs of thyrotoxicosis. Thyroid bruit was absent. Pulse rate was 96/minute, blood pressure of 140/80mmHg, and respiratory rate 18/minute. On central nervous system examination, the higher mental functions and cranial nerve examination were within normal limits. Motor system examination showed the presence of flaccid quadriparesis with areflexia. Sensory system examination was within normal limits. Cardiovascular and respiratory system examination were normal.
Investigations revealed: haemoglobin (Hb) -13.1 gm%, total count (TC) - 11,400/cmm, platelet count - 49,000/cmm, random blood sugar (RBS) - 110mg/dl, blood urea - 29 mg/dl, serum creatinine - 0.8 mg/dl. Serum electrolyte profile showed sodium - 143 mEq/L, potassium - 2.2mEq/L, chloride - 112mEq/L. Serum calcium and magnesium levels were within normal limits. Electrocardiogram (ECG) was normal. Human Immunodeficiency Virus (HIV) ELISA was non reactive. Bone marrow biopsy and ultrasonography of abdomen were normal. Fine Needle Aspiration Cytology (FNAC) of thyroid showed features of hyperplastic colloid goitre. Ultrasonography of thyroid showed hyperechoic small nodules in both lobes as well as isthmus suggestive of multinodular goitre. Thyroid profile was: total T3 - 2.34 (normal: 0.60 - 1.81ng/ml), total T4 - 13.9 (normal: 4.5 - 10.9 mcg/dl), thyroid-stimulating hormone (TSH) - 0.01 (normal: 0.35 - 5.5IU/ml). Antithyroid antibodies and antiplatelet antibodies were negative. Nerve conduction study was normal. A final diagnosis of TPP with idiopathic thrombocytopenia was made.
The patient was administered 40mmol potassium chloride intravenously. He was treated with tablet carbimazole 10mg three times a day and tablet propanolol 10mg twice a day. The patient’s weakness in all four limbs improved dramatically within an hour after potassium chloride administration. As he had persistent thrombocytopenia during his stay in hospital, he was commenced on tablet prednisolone (1mg/kg body weight). His platelet count normalized in one month after which the steroid dose was tapered and stopped.
Discussion
TPP is an uncommon disorder characterised by simultaneous thyrotoxicosis, hypokalaemia and paralysis that occurs primarily in males of South Asian descent. The overall incidence of TPP in Chinese and Japanese thyrotoxic patients is 1.8% and 1.9% respectively.3, 4 Sporadic cases have been reported in non-Asian populations such as Caucasians, Afro-Americans, American Indians and Hispanics. With population mobility and admixture, TPP is becoming more common in Western countries. Many affected patients are in the age group of 20 - 40 years and do not have obvious symptoms and signs of hyperthyroidism.5 The attack is characterised by recurrent, transient episodes of muscle weakness that range from mild weakness to complete flaccid paralysis. The proximal muscles are affected more severely than distal muscles. Attacks usually first involve the lower limbs, and progress to the girdle muscles and subsequently the upper limbs. Sensory function is not affected. Although patients can present with quadriparesis that resembles Guillain-Barre Syndrome or transverse myelitis, the bladder and bowel functions are never affected. Patient may experience recurrent episodes of weakness that last from a few hours up to 72 hours with complete recovery between the attacks. In the majority of patients, deep tendon jerks are markedly diminished or absent although some patients may have normal jerks.
Patients with TPP usually experience the attacks a few hours after a heavy meal or in the early morning hours upon waking. More than two-thirds present to the emergency department between 2100 and 0900 hours; hence it was initially described as nocturnal palsy or night palsy.6 It has been shown that plasma glucose and insulin responses to meals are markedly higher in the evening than in the morning in control subjects. Such a phenomenon suggests a possible mechanism for the nocturnal preponderance of TPP. Another explanation could be the circadian rhythmicity of many hormones reaching their peak levels during sleep. Hypokalaemia is considered to be the most consistent electrolyte abnormality in TPP and a hallmark of the syndrome along with hyperthyroidism. It has been demonstrated that hypokalaemia is a result of potassium shift into cells and that it is not caused by total body potassium depletion.7 Patients with thyrotoxic periodic paralysis have an underlying predisposition for activation of Na+/K+-ATPase activity either directly by thyroid hormones or indirectly via adrenergic stimulation, insulin or exercise. Increased Na+-K+ ATPase activity is postulated to contribute to hypokalaemia.8
The majority of cases of hyperthyroidism associated with thyrotoxic periodic paralysis are due to Graves disease although other conditions including thyroiditis, toxic multinodular goitre, toxic adenoma, TSH secreting pituitary tumour, ingestion of T4 and inadvertent iodine excess have also been implicated.9 Assaying of thyroid function in patients with hypokalaemic paralysis distinguishes thyrotoxic periodic paralysis from other forms of hypokalaemic periodic paralysis. Thyrotoxic periodic paralysis occurs only in the presence of hyperthyroidism and is abolished when thyroid hormones are normalised.
Immediate therapy with potassium supplementation and beta-adrenergic blockers can prevent serious cardiopulmonary complications and may hasten recovery of periodic paralysis.10 Potassium chloride is given intravenously and/or orally. Regular potassium supplementation as prophylaxis against further paralysis when the patient has normal serum potassium level is ineffective. Effective control of hyperthyroidism is indicated to prevent recurrence of paralysis.
Conclusion
To conclude, although the association of thyrotoxicosis and periodic paralysis has been well known, TPP is often not recognised when first seen because of lack of familiarity with the disorder and partly because of the subtleness of thyrotoxicosis. When a young male of South Asian descent is initially seen with severe lower limb weakness or paralysis, TPP should be considered in the differential diagnosis and investigated for its presence since it is a curable disorder that resolves when euthyroid state is achieved.
William Osler has said that "A desire to take medicine is perhaps the great feature which distinguishes man from animals" This desire, however may play havoc when a person starts taking medicines on their own (i.e. self-medicating), forgetting that all drugs are toxic and their justifiable use in therapy is based on a calculable risk 1.
Self-medication (SM) can be defined as obtaining and consuming drugs without the advice of a physician2. There is a lot of public and professional concern about the irrational use of drugs in SM. In developing countries like India, easy availability of a wide range of drugs coupled with inadequate health services result in increased proportions of drugs used as SM compared to prescribed drugs2. Although, over-the-counter (OTC) drugs are meant for SM and are of proved efficacy and safety, their improper use due to lack of knowledge of their side effects and interactions could have serious implications, especially in extremes of ages (children and old age) and special physiological conditions like pregnancy and lactation 3, 4. There is always a risk of interaction between active ingredients of hidden preparations of OTC drugs and prescription medicines, as well as increased risk of worsening of existing disease pathology 5 . As very few studies have been published in our community regarding usage of self medication we conducted this cross-sectional study in the coastal region of Pudhucherry, South India, t assess the prevalence and pattern of SM use.
Materials and methods:
The present study was a cross-sectional survey conducted in coastal region of pudhucherry, south India. For this study we recruited 200 patients randomly from both urban and rural communities (100 each) for a period of six months during 2009. Patients who were = 18 years of age and who were able to read and write the local language (Tamil) or English were included in the study after informed consent explaining the purpose of the study. Participants with intellectual, psychiatric and emotional disturbances that could affect the reliability of their responses were excluded from the study. To collect data regarding SM usage a structured questionnaire was prepared, after an extensive literature review.. The structured questionnaire contained 25 items in the form of closed and open ended questions. Initially the tool was validated by a panel of experts in the field of public health for the appropriateness of each item and assessment of content validity (0.91) and re-test reliability coefficient (0.89). Approval to conduct the study was granted by the Institute ethics committee prior to data collection. Each participant underwent a face to face interview to collect data followed by an informal educational counseling about potential adverse effects of consuming common SM. Data collected was analyzed using SPSS for windows statistical software version 14 (SPSS Inc., Chicago, Il, USA). Data was presented using descriptive statistics (i.e. numbers, percentage) and inferential statistics (i.e. Chi-square). A probability value of < 0.05 was considered to be significant.
Results
Basic demographic details:
The majority of the participants were female (56%). Most of the participants (60%) were between 26-45 years of age. There were an equal number of participants from the rural and urban community. Among the total 200 participants 70% were literate.
Findings related to usage of SM:
Overall, out of 200 participants, 71 % of them reported that they have used SM in the past. The frequency of SM use varied among the subjects with a minimum of at least one time to maximum of 5 times and above See Figure 1. When the participants were asked about the reasons for SM use, the majority of them - 41.5% - stated lack of time to visit a doctor as the main reason followed by minor illness and quick relief. See Table 1. The major source through which the participants learned to use SM were as follows, directly from pharmacist (57.3%), prescription of previous illness (21.5%), friends (12.5%), television (5.5%) and books (3%).See Table 2. The main indications for SM use were fever (36%), headache (35%), then cough/cold/sore throat (20%). See Table 3 for detailed data.
Figure 1: Frequency of self medication Use
Table 1: Reasons for Self Medication Use
Reasons
Number (%)
Lack of time
41.5
Minor illness
10.5
Economical
14
Quick relief
10
Learning opportunity
2
Ease and convenience
10.5
Avoiding crowd in visiting doctor
6
Unavailability of doctor
5.5
Table 2: Sources of Self Medication Use
Sources for self medication use
Number (%)
Directly from pharmacy without prescription
57.3
Prescription of previous illness
21.5
Friends prescription
12.5
Television media
5.5
Book
3
Table 3: Indications for Self Medication Use
Indications for self medication use
Number (%)
Headache
35
Stomach ache
3
Vomiting
1
Eye symptoms
0.73
Diarrhoea
2
Cough, cold, sore throat
20
Fever
36
Skin symptoms
0.27
Ear symptoms
2
While calculating chi-square to find out the association between usage of SM and selected demographic variables we found an association between residence (i.e. rural or urban) and gender; urban people were more likely to use SM than rural people (urban, 60/100 vs. rural 82/100, p value = .006). In relation to gender females were more likely to use SM in comparison to males (female, 78/112 vs. 43/88, p value= .002). Other variables were not significantly associated with SM use. Finally, when the subjects were asked about the side effects of their used self medications 93.5% of them said that they are not aware of the side effects and only the remaining 6.5% of them said they are aware of the side effects.
Discussion
The current study examined the prevalence and pattern of SM use in a coastal region of South India. The study findings revealed 71% of the people reporting SM use in the past, this prevalence rate in our study is consistent with previous finding3,6,7,8,9,10,11 The figure of participants who use SM is very high, which requires immediate attention. The frequency of self medication use in our study ranged from a minimum of one time to a maximum of 5 times and above, this finding was in line with the findings of a study by Nalini (2010)12.
Participants cited multiple reasons for use of SM like lack of time , quick relief from illness and ease and convenience, a similar reasons were cited in an another Indian study13. In the current study participants reported SM use in a variety of conditions like headache, stomach ache, cough and fever, this these finding are comparable with those of Sontakke et al (2011) 14. The reason for SM use may be mufti-factorial, in our study an association was found between gender and residence, i.e. female and rural people reporting more SM use, this finding was similar to two previous studies15,16 To establish the reasons why requires further research. One potential limitation of this study is the limited sample size, which we tried to overcome by adopting a random sampling method so as to generalize findings.
Conclusion
Factors influencing SM include patient satisfaction with the healthcare provider, cost of the drugs, educational level, socioeconomic factors, age and gender 17. Interactions between prescribed drugs and the drugs taken for SM is an important risk factor of which healthcare providers must be aware of.17,2
Easy availability of wide range of drugs without a prescription is the major factor responsible for irrational use of drugs in SM as, thus resulting in impending health problems (antimicrobial resistance, increased load of mortality and morbidity) and economic loss. The need for promoting appropriate use of drugs in the health care system is not only for financial reasons, with which policy makers and manager are usually most concerned, but also for health and medical care of patients and the community. There is need for authorities to strengthen existing laws regarding OTC drugs to ensure their rational sale and use. Also, specific pharmacovigilance is needed and the patient, pharmacist and physician must be encouraged to report any adverse events. Periodic studies on the knowledge, attitude about and practice of SM may give insight into the changing pattern of drug use in societies.
An 80-year old lady was referred to a gastroenterology clinic in August 2009 with deranged liver function tests; alkaline phosphatase 180 IU/L (35-120), alanine transferase 147 IU/L (<40), gamma glutamyl transferase 384 IU/L (<45) and globulins 45 g/L (20-35). She had initially presented to her general practitioner with symptoms of lethargy and malaise four months previously. She denied any symptoms of obstructive jaundice and there were no risk factors for hepatitis; she seldom consumed alcohol.
Past medical history included osteoarthritis, migraines and recurrent urinary tract infections; these had been investigated by urology and the patient had undergone cystoscopy and urethral dilatation in September 2003; despite this she continued to experience urinary tract infections and was therefore commenced on prophylactic nitrofurantion by her General Practitioner with approval by the Urologist. This was initially commenced at 50mg at night. This regime was continued for approximately three years however during this time she had a further three treatment courses of nitrofurantoin. In October 2005 her prophylactic dose was therefore increased to 100mg at night. Other medication included lansoprazole 30mg daily, pizotifen 500 micrograms at night, metoprolol 100mg twice daily, simvastatin 10mg at night, senna 15mg at night and furosemide 40mg daily.
On examination there was evidence of palmar erythema and Dupuytren’s contractures but no other stigmata of chronic liver disease. The liver was tender and palpable 4 cm below the costal margin. A liver ultrasound was performed which was normal. Liver screen and autoimmune profile are shown in table 1; notably a positive nuclear antibody was found (1 in 1280 IgG) with Hep 2 cell staining showing a homogenous (ANA) pattern at 1:320 IgG, and a nuclear lamin pattern at 1:1280 IgG;. Due to the positive ANA and raised globulins a suspected diagnosis of nitrofurantoin-induced autoimmune hepatitis was made and a liver biopsy performed.
Liver biopsy (figure 1) indicated a moderate hepatitis which was mainly portal based with multifocal interface hepatitis; these morphological appearances were consistent with those of an autoimmune hepatitis. The patient was advised to immediately stop nitrofurantoin and was commenced on prednisolone 30mg which caused a rapid improvement in LFTs (figure 2). This improvement was maintained following a step-wise reduction in steroid dose and prednisolone was discontinued after eight months of treatment. LFTs are currently normal one month following cessation of steroids
Discussion
This case raises two points of discussion. The first is whether the long term use of nitrofurantoin as prophylaxis for urinary tract infections is appropriate and based on solid evidence. Nitrofurantoin has many side effects and is well documented to cause liver derangement1,2,3. The patient described in this case had been taking nitrofurantoin for seven years and had received a large cumulative dose, on the basis that this was effective prophylaxis. The continuous, long term use of antibiotics as prophylaxis for urinary tract infections is debatable. Madersbacher et al4 recommend the use of prophylactic antibiotics but only after or alongside additional measures including behavioural change, the use of topical oestrogens and the use of alternative therapies; this view is supported by the European Association of Urology5.A Cochrane Review6 in 2004 found that antibiotic use did decrease the number of urinary tract infections compared to placebo but only for the duration of treatment; antibiotics do not alter the natural history of the underlying condition7. There is no clear evidence for duration of treatment and any trials have only been continued for six or twelve months6. It has been noted that all antibiotics had a worse adverse event profile compared to placebo. There was no consensus as to which antibiotic should be used although nitrofurantoin has been associated with a greater withdrawal rate6. One study8 comparing nitrofurantoin and trimethoprim revealed no significant difference in recurrence rates or side effects between the two antibiotics, although this involved a lower dose of nitrofurantoin than was used in this case, and a treatment duration of just 6 months. We would argue that due to the side effect profile of nitrofurantoin and the evidence base available, it is not appropriate to continue it for a duration beyond 6 months.
The second discussion point is whether nitrofurantoin was actually the cause for liver derangement in this case. As documented in a recent review article on the diagnosis of drug-induced liver injury, establishing with any certainty whether liver injury is drug induced can be very difficult3. The key issues are whether there is a temporal relationship between the drug and the onset of liver injury, and whether other causes have been excluded. In this case the patient had negative viral serology and a normal ferritin and caeruloplasmin but her positive autoantibodies raise the possibility of autoimmune hepatitis. Guidelines from the American Association for Liver Diseases9 suggests that the diagnosis of autoimmune hepatitis should be made on the following criteria
laboratory abnormalities (serum AST or ALT, and increased serum total IgG or gamma-globulins)
positive serological markers including ANA,SMA, anti-LKM1 or anti- LC1
histological changes consistent with autoimmune hepatitis i.e. interface hepatitis
This case meets these above criteria for autoimmune hepatitis however the presence of nitrofurantoin does confound the issue. Other case reports10 have reported nitrofurantoin to have caused autoimmune hepatitis based on the relationship between the timing of the drug and the onset of biochemical abnormalities. Bjornsson et al11 performed a comparative study of patients with autoimmune hepatitis and found drugs, particularly nitrofurantoin and minocycline were causally implicated in 9% of cases. When they compared the two groups no significant differences were found in the diagnostic parameters of biochemical, serological and histological abnormalities. In fact the only difference was that no drug-induced cases relapsed on withdrawal of steroids whereas nearly two third of those with non-drug-induced hepatitis relapsed. Bjornsson et al therefore argue in favour of autoimmune immune hepatitis being induced by drugs such as nitrofurantoin; rather than particular drugs simply unmasking sporadic cases based on these management differences.
The patient in this case so far has shown no signs of relapse following steroid withdrawal. We believe that this case does represent one of nitrofurantoin-induced autoimmune hepatitis. In view of the above we would urge readers to consider their use of nitrofurantoin for recurrent urinary-tract infection prophylaxis.
Chronic obstructive pulmonary disease (COPD) is a debilitating condition resulting in significant morbidity and mortality. It is the fifth leading cause of death in the UK 1, estimated to be the third by 2020 2.
Definition:
COPD is a preventable and treatable disease with some extra-pulmonary effects that may contribute to the severity in individual patients Its pulmonary component is characterised by airflow limitation that is progressive and not fully reversible. There is an abnormal inflammatory response of the lung to noxious gases and particles, most commonly cigarette smoke 3.
Airflow obstruction is defined as post-bronchodilator FEV1/FVC ratio (where FEV1 is the forced expiratory volume in one second and FVC is the forced vital capacity) of less than 0.7 If FEV1 is ≥ 80% predicted, a diagnosis of COPD should only be made in the presence of respiratory symptoms 4.
Incidence/ Prevalence:
Within the UK it is estimated that 3 million people are affected with COPD 4. However, only 900,000 are diagnosed -4An estimated two million people who have COPD remain undiagnosed 4.
Causes:
90% of cases are smoking related 4, particularly those with >20 pack year smoking histories 5. Environmental and occupational factors can also play a role, including exposure to biomass fuels such as: coal, straw, animal dung, wood and crop residue which are used to cook in some countries and heat poorly ventilated homes COPD occurs in 10-20% of smokers, suggesting there is an element of genetic susceptibility 2-3, 5.
Diagnosis:
To make a diagnosis of COPD an obstructive deficit must be demonstrated on spirometry in patients over the age of 35 years with risk factors (mainly smoking) and signs and symptoms of the disease 4.
Signs and Symptoms:
Progressive dyspnoea on exertion
Chronic cough
Chronic sputum production
Wheeze
Frequency of exacerbations – particularly during winter months 4
Functional status – bearing in mind gradual progression of disability, effort intolerance and fatigue.
Features suggestive of Cor pulmonale 5:
Peripheral oedema
Elevated jugular venous pressure
Hepatomegaly
Right ventricular heave
Tricuspid regurgitation
Investigations/ Tests to consider:
Post-bronchodilator Spirometry – essential in confirming the diagnosis of COPD.
Demonstrating an obstructive picture.
FEV1 is used to assess the progression and severity of COPD, but correlates poorly with the degree of dyspnoea 3-6. (Table 1)
Pulmonary functions tests – Markers suggesting the presence of emphysema include:
Reduced TLCO and KCO due to a reduced surface area for gaseous exchange 5.
Raised Total lung capacity, residual volume and functional residual capacity due to air trapping 5.
Chest radiograph – Is not required for the diagnosis, but is recommended to exclude other conditions such as interstitial lung disease, pleural effusions or pneumothorax. It may demonstrate features of the condition, such as 3, 5:
Hyperinflated lung fields
Flattened diaphragms
Bullous changes, particularly at the apices
BODE index prognostic indicator – This is grading system shown to be better than FEV1 at predicting the risk of hospitalisation and death in patients with COPD. Patients are scored between 0 and 10, with higher scores having an increased risk of death. It encompasses 3, 5-7: (Table 2)
BMI
Airflow Obstruction – taking into account the FEV1
Dyspnoea – in accordance with the Medical Research Council (MRC) scale 5.
Exercise capacity – measured by the distance walked in 6 minutes. (Table 3)
Table 1. Severity of airflow obstruction 4
Stage
Severity post-bronchodilator
FEV1 (%) Predicted
Comments
1
Mild
≥ 80%
Only diagnosed in the presence of symptoms
2
Moderate
50- 79%
Managed within the community
3
Severe
30-49%
TLCO usually Low
Hospitalization may be needed only with exacerbations
4
Very Severe
<30%
Or FEV1 <50% with respiratory failure
Table 2. BODE Index 3, 5-8
1
2
3
FEV1 Predicted (%)
≥ 65
50- 64
36- 49
≤ 35
Distance walked in 6 minutes (meters)
≥ 350
250- 349
150- 249
≤ 149
MRC dyspnoea scale
0-1
2
3
4
BMI
≥ 21
≤ 21
Table 3. Medical research council (MRC) Dyspnoea scale 5, 8
1
Dyspnoeic only on strenuous activity
2
Dyspnoeic on walking up a slight incline or when hurrying
3
Walks slower than contemporaries on the flat, or has to stop for breath, or has to stop for breath when walking at own pace
4
Stops for breath on walking 100m or after a few minutes on walking on the flat
5
Breathless on minimal exertion e.g. dressing/ undressing. To breathless to leave the house
Differential Diagnosis:
Asthma – the most important differential diagnosis to consider.
This is steroid and bronchodilator responsive
Indicative of reversible airway obstruction.
It is not associated with smoking.
Patients with asthma may exhibit 3, 9: chronic non-productive cough, variability in breathlessness, diurnal /day-to-day variation, nocturnal wheeze and dyspnoea
However both conditions may coexist creating diagnostic uncertainty.
Alpha1 antitrypsin deficiency is an autosomal dominant condition associated with an increased risk of developing emphysema at an early age 3, 5, 9.
It can occur in non-smokers
Can be asymptomatic and thus under-diagnosed with an estimated 1 in 2000-5000 individuals being affected 5.
The disease is worse in smokers
COPD can develop in patients < 35years of age
It is associated with liver cirrhosis.
All patients with COPD should be screened.
Emphasis should be made to avoid smoking, including passive smoking.
Other conditions to consider include:
Bronchiectasis
Interstitial lung disease
Cardiac failure.
Treatment:
Goals of management include:
Early and accurate diagnosis
Improve symptoms and quality of life
Reduce the number of exacerbations
Improve mortality
Non-pharmacological management:
Smoking cessation – an accurate smoking history should be obtained, including the number of pack years smoked. All current smokers with COPD should be encouraged to stop at every opportunity, and offered smoking cessation advice. Advising the patient alone will help a certain proportion to stop, whilst referral to smoking cessation services has been shown to further increase in quit rates. There are a range of nicotine and other pharmacological therapies available such as Bupropion (Zyban®) and Varenicline (Champix®) 3-4, 7, 8.
Vaccinations – A once off Pneumococcal and annual Influenza vaccine should be offered.
Pulmonary rehabilitation – Should be offered to patients who have had a recent exacerbation requiring hospitalisation and those that have an MRC score of ≥ 3, but are still able to mobilise and thus have the potential for further rehabilitation. It is not suitable for those patients that are immobile or limited in their mobility due to symptoms of unstable angina or a recent cardiac event. Benefits are seen in terms of reduced hospital admission, improved quality of life and exercise tolerance Commitment to the programme should be relayed to the patient, and each programme should be tailored to their individual needs. This usually includes 3-5:
Disease education – which can improve the ability to manage their illness.
Exercise – tailored programmes to prevent de-conditioning and improve functional exercise capacity, dyspnoea and quality of life 4. This includes strength and endurance training of upper limbs and respiratory muscles Benefits may be seen even after 6 months.
Physiotherapy – to teach active cycle breathing techniques or to use positive expiratory pressure masks in patients with excessive sputum production.
Nutritional support – in the form of supplementation or dietician advice in patients with a suboptimal BMI. A low BMI is associated with increased mortality as it is associated with poor exercise capacity, reduced diaphragmatic mass and impaired pulmonary status. Alternatively, weight loss is recommended in patients who are in the obese range.
Psychological – Assessment for support at home, introduction of patients to day centres, assessing for features of depression and anxiety, and aiding in the obtainment of a car disability badges may require referral to occupational therapy and social services.
Travel advice – Patients who are planning air travel and have FEV1 <50%, Sa02 < 93%, or are on long term oxygen therapy (LTOT) should undergo formal assessment -4Patients with bullous disease should be informed that they are at increased risk of pneumothorax during high altitude flights 4.
Pharmacological management:
Bronchodilators – Provide long term benefit in reducing dyspnoea. This is not reflected in improvements in FEV1 as it may not show reversibility 4.
Start with an inhaled SABA (short-acting beta2-agonist) or a SAMA (short-acting muscarinic antagonist) on an as required basis for symptomatic relief. If symptoms remain despite regular SABA therapy (i.e. four times a day), then treatment will need to be stepped up.
If symptoms persist or if the patient is having recurrent exacerbations add in a LABA (long-acting beta2 agonist) or a LAMA (long acting muscarinic antagonist).
If symptoms continue, add in a LAMA if already on a LABA (or vice versa).
If FEV1 <50% add in an inhaled corticosteroid (ICS). This can be offered as a combination inhaler.
Inhaled therapy should offer sufficient bronchodilator response. A spacer can be used for those with poor technique. Nebulisers are reserved for patients who demonstrate respiratory distress despite maximal inhaled therapy, and for those that show an improvement in symptoms or exertional capacity 4.
Diagram 1: Summary of step-by-step management 4
Corticosteroids – A short course of oral steroids may be used during exacerbations. A maintenance course however is not recommended Any patients on long term steroids should be weaned off.
Mucolytic agents – May be considered in patients with a chronic cough who have difficulty expectorating. They should only be continued if symptomatic benefit is evident, otherwise they can be stopped. There is no evidence to show that they reduce the exacerbation frequency.
Theophylline – Should only be offered in people that are unable to use inhaled therapy or after trials of SA and LA bronchodilators 4. The same generic brand should be prescribed as individual brands will have different efficacy. It is usually used as an adjunct to beta2-agonists and muscarinic antagonists. Interactions with macrolides and fluroquinolones and other drugs are also common, and as such the theophylline dose should be reduced if interactions are known. Caution should be taken in prescribing theophylline in the polypharmacy patient 3, 5. Little evidence has been shown to support theophylline usage in COPD (compared to asthma), however it is used for its anti-inflammatory effects As such levels are only performed if toxicity is suspected and should not be adjusted if in the sub-therapeutic range.
Oxygen therapy – Patients should be assessed for long-term oxygen therapy(LTOT) if they exhibit 4:
Severe airflow obstruction
Features of Cor pulmonale
Hypoxaemia (Sa02 ≤ 90%)
Cyanosis
Polycythaemia
Patients with stable COPD who are receiving maximum medical therapy are assessed by measuring arterial blood gases taken on two separate occasions at least 3 weeks apart. To meet the criteria patients must have 4:
A Pa02 < 7.3 kPa when stable, or
A Pa02 >7.3 but < 8.0 kPa when stable and:
Pulmonary hypertension or
Peripheral oedema or
Secondary polycythaemia or
Nocturnal hypoxaemia
LTOT should be used for a minimum of 15L per day, including during sleep 3-4.
Patients who continue to smoke should be made aware of the serious risk of facial injuries due to the highly flammable nature of oxygen.
When to refer:
Referrals for specialist advice or specialist investigations may be appropriate at any stage of the disease.
Other possible reasons for referral 4
§ Diagnostic uncertainty
§ Suspected severe COPD
§ Onset of Cor pulmonale
§ Rapid decline in FEV1
§ Assessment for LTOT, home nebulisers or oral corticosteroid therapy
§ Symptoms that do not correlate to lung function deficit
§ Pulmonary rehabilitation assessment
§ Frequent infective exacerbations
§ Family history of alpha-1-antitrypsin deficiency
§ Haemoptysis
§ Onset of symptoms < 40 years
§ Bullous lung disease
§ Assessment for lung volume reduction surgery/ lung transplantation
§ Dysfunctional breathing
Follow-up:
Patients with stable mild-moderate COPD should be reviewed by their general practitioner at least once a year and those with severe COPD twice yearly.
At each visit 4:
An opportunity should be taken to ask about their current smoking status and the desire to stop.
Assessment of adequate control of symptom: dyspnoea, exercise tolerance and the estimated number of exacerbations per year.
Assessment of inhaler technique.
To assess the effects/side effects of each drug treatment.
The need for pulmonary rehabilitation.
For those patients with very severe airflow obstruction (FEV1 < 30%), the above still remains, in addition to the assessment of 4:
Features of Cor pulmonale
Nutritional status
The need for LTOT
Signs of depression
The need for occupational therapy and social services input
Referral to specialist and their services
Measurements of:
FEV1 and FVC
BMI
MRC dyspnoea scale
Sa02 via pulse oximetry
Those patients requiring long term non-invasive ventilation will be reviewed by a specialist on a regular basis.
Warfarin is the most commonly used oral anticoagulant and has established efficacy for more than 50 years for the prevention of thromboembolic events, but its use is limited by fear of bleeding, drug-drug and drug-food interactions, and routine monitoring of international normalized ratio (INR). In patients with atrial fibrillation (AF), warfarin prevents 64% of strokes in research studies but the real-world effectiveness drops to 35% because of various factors leading to its suboptimal use.1 In October 2010 the United States (US) Food and Drug Administration (FDA) approved Pradaxa capsules (dabigatran etexilate) as the first new agent to prevent stroke and systemic emboli in patients with non-valvular AF. In this article we will discuss some of the evidence for and against the use of dabigatran.
In the RE-LY study2 (Randomized Evaluation of Long-term Anticoagulant Therapy), high-dose dabigatran (150mg twice a day) was found to be superior to warfarin for the prevention of stroke and systemic emboli, required no routine INR monitoring, and had few food and drug interactions. James Freeman and colleagues,3 using data from the RE-LY trial, found that high-dose dabigatran (150mg twice a day) was the most efficacious and cost-effective strategy compared with adjusted-dose warfarin among adults older than 65 with AF.
Dabigatran has been shown to specifically and reversibly inhibit thrombin, the key enzyme in the coagulation cascade. Studies in healthy volunteers4 and in patients undergoing orthopaedic surgery have indicated that dabigatran has a predictable pharmacokinetic/pharmacodynamic profile, allowing for a fixed-dose regimen. Peak plasma concentrations of dabigatran are reached approximately two hours after oral administration in healthy volunteers, with no unexpected accumulation of drug concentrations upon multiple dosing. Excretion is predominantly via the renal route as unchanged drug. Dabigatran is not metabolized by cytochrome P450 isoenzymes. Though use of dabigatran for non-valvular AF and venous thromboembolism (VTE) is gaining practice,5 it remains far from being the standard of care.
What are the concerns with use of dabigatran? In the RE-LY study the INR control was relatively poor (64% TTR (time in the therapeutic range)) but, probably more importantly, the relationship between events and individual’s INR control was not reported. The use of centre’s time in therapeutic range (cTTR) in the RE-LY study as a surrogate for INR control may not truly reflect TTRs for individual patients. Also in RE-LY study, randomization was stratified for centre and by the centre-based analyses, and the quality of oral anticoagulant services was the basis for the comparisons in this report. A subgroup analysis6 concluded that relative effectiveness of dabigatran versus warfarin was mainly seen at centres with poorer INR control. For example, Swedish centres had good TTR and the relative effectiveness and safety of dabigatran was virtually the same as with warfarin; thus, it is only the price difference that counts. It also highlights how local standards of care affect the benefits of use of new treatment alternatives and hence further limits the generalizability of any ‘overall average’ cost-effectiveness of dabigatran, raising the question that if an intervention does not do more, why should a payer pay more for it? There are several other factors that could impact on the cost-effectiveness7 of dabigatran such as patient medication adherence, dosing frequency, and the potential effect of new efficient methods of warfarin management improving INR control by patient self-testing.
The other shortcomings of dabigatran include lack of antidotes when patients do bleed and lack of any alert to physicians that patients are not compliant with dabigatran (INR serves this purpose for warfarin). Additionally, in the RE-LY trial, dabigatran was used twice daily thus raising compliance issues compared to once daily warfarin (the rates of discontinuation of dabigatran were higher at 15% and 21% at one and two years, respectively); 11.3% reported dyspepsia (twice the rate of warfarin group); high rate of gastrointestinal bleed compared with warfarin; patients in the dabigatran cohort were at slightly higher risk of myocardial infarction (not sure how it will translate in real world practice); and contraindication of dabigatran in severe renal dysfunction raises some more questions about its use and cost effectiveness. In addition, the RE-LY trial excluded patients who had: contraindications to anticoagulation, severe heart-valve disorder, stroke within 14 days or severe stroke within six months before screening, a condition that increased risk of haemorrhage, creatinine clearance of less than 30ml per minute, active liver disease, and pregnancy. Clinicians will need to use their judgement to weigh and balance the risk for bleeding with this new agent in a setting of an acute stroke versus the risk of having another ischaemic stroke in someone with AF if not given anti-coagulation therapy immediately. Safety and efficacy at extremes of body weight is not well established with current FDA approved doses of dabigatran either.
In summary dabigatran is a very exciting new agent with significant advantages over warfarin. However, in view of dabigatran’s higher non-adherence rate and greater risk of non-haemorrhagic side effects, patients already taking warfarin with excellent INR control have little to gain by switching to dabigatran.1 Until more studies and post-marketing data become widely available, we should advocate tight INR control for which there is a wealth of evidence for benefits, and promote strategies to improve the management of therapy with warfarin.
Lactic acidosis is an important cause of metabolic acidosis in hospitalised patients. This usually occurs either due to over production or under utilisation of lactate1 . Most cases of lactic acidosis are due to marked tissue hypoperfusion or hypoxia in systemic shock.
Asymptomatic lactic acidosis has been reported previously during acute severe asthma and attributed to fatiguing respiratory muscles, hypoxaemia and liver ischaemia. It has also been linked to β2 agonist therapy in asthma, although lactic acidosis causing increasing dyspnoea in the asthmatic patient has only been recorded rarely.
Case presentation
We present a case of lactic acidosis in a patient with acute severe asthma who did not have any overt signs of sepsis or tissue hypoperfusion.
Mr IL was a 49 years old male who was known to have moderate asthma. He had multiple previous admissions to hospital with exacerbation of asthma but had never required an intensive care admission and had never been intubated. His other comorbidities included atrial fibrillation, ischaemic heart disease and depression.
His usual medications included salbutamol, budesonide and salmeterol inhalers, aspirin, atorvastatin and digoxin. He was a mechanic by trade with no obvious occupational sensitisation. He had no pets at home. He was a smoker with a 20 pack year history. Recent lung function tests showed an FEV1/FVC of 0.68 with a post bronchodilator FEV1 of 4.17 L (95% predicted).
He was admitted with a 1 week history of worsening shortness of breath, dry cough and wheeze. His baseline blood tests including full blood count, C reactive protein, liver and renal function were normal. Chest radiograph was unremarkable. Arterial blood gas showed no evidence of hypoxia or acidosis.He was treated as acute severe asthma with back to back nebulisers, intravenous hydrocortisone and magnesium sulphate resulting in gradual improvement in bronchospasm and peak expiratory flow rate.
Despite optimal treatment, his breathing started to deteriorate. Arterial blood gas at this time showed lactic acidosis with normal oxygenation (Table 1). There was no clinical or biochemical evidence of haemodynamic compromise or sepsis. A presumptive diagnosis of lactic acidosis secondary to salbutamol was made. The nebulisers were withheld and he has transferred to high dependency unit for closer monitoring. The acidosis completely resolved in the following 12 hours on stopping salbutamol and the patient made an uneventful recovery.
Table 1: Serial Arterial Blood Gases (On admission, 4 hours later and on stopping salbutamol)
00:22
04:06
07:42 *
10:50
11:35
12:24
14:29
17:33
23:32
FiO2
100%
60%
60%
60%
60%
40%
40%
35%
28%
pH (7.35-7.45)
7.36
7.28
7.26
7.32
7.34
7.37
7.37
7.39
7.41
pCO2 (4.5-6.0 kPa)
4.87
4.74
4.15
3.31
3.98
3.9
4.7
5.08
5.49
pO2 (11-14 kPa)
27
19.2
16.5
19
18
14.1
12.5
13
11.8
HCO3 (22-28 mmol/L)
22
16.3
13.6
12.4
15.6
16.6
19.9
22
25.6
BE (2- -2)
-2
-9.1
-12
-12
-9
-7.6
-4.4
-1.5
1.4
Lactate (0.5-2 mEq/L)
1.8
7.6
9.7
9.3
7.6
6.8
3.6
1.4
1.1
* Salbutamol witheld
Discussion
Lactate is a product of anaerobic glucose metabolism and is generated from pyruvate. Normal plasma lactate concentration is 0.5-2 meq/L. Most cases of lactic acidosis are due to marked tissue hypoperfusion or hypoxia in systemic shock2 .
Lactic acidosis can occur in acute severe asthma due to inadequate oxygen delivery to the respiratory muscles to meet an elevated oxygen demand3 or due to fatiguing respiratory muscles4 . A less recognised cause of lactic acidosis is treatment with salbutamol. The mechanism of this complication is poorly understood.
Salbutamol is the most commonly used short acting βagonist. Stimulation of β adrenergic receptors leads to a variety of metabolic effects including increase in glycogenolysis, gluconeogenesis and lipolysis5 thus contributing to lactic acidosis.
Table 2 shows an assortment of previously published case reports and case series of lactic acidosis in the context of acute asthma.
Table 2: Details of etiology and consequences of lactic acidosis in previously published case reports
Reference
n
Suggested etiology of lactic acidosis
Effect of lactic acidosis
Roncoroni et al, 1976 [6]
25
Uncertain: increased respiratory muscle production, decreased muscle or liver metabolism
None observed
Appel et al, 1983 [7]
12
Increased respiratory muscle production, decreased muscle or liver metabolism
8 out of 12 developed respiratory acidosis, 6 required invasive ventilation
Braden et al, 1985 [8]
1
β2 agonist, steroid and theophylline therapy
None
O’Connell & Iber, 1990 [9]
3
Uncertain: intravenous β2 agonist versus severe asthma
None
Mountain et al, 1990 [10]
27
Hypoxia and increased respiratory muscle production
None
Maury et al, 1997 [11]
1
β2 agonist therapy
Inappropriate intensification of β2 agonist therapy
Prakash and Mehta, 2001 [2]
2
β2 agonist therapy
Contributed to hypercapneic respiratory failure
Manthous, 2001 [12]
3
β2 agonist therapy
None
Stratakos et al, 2002 [3]
5
β2 agonist therapy
None
Creagh-Brown and Ball, 2008 [13]
1
β2 agonist therapy
Patient required invasive ventilation
Veenith and Pearce, 2008 [14]
1
β2 agonist therapy
None
Saxena and Marais, 2010 [15]
1
β2 agonist therapy
None
Conclusion
In this case, the patient developed lactic acidosis secondary to treatment with salbutamol nebulisers. The acidosis resolved spontaneously without any specific treatment.
Lactic acidosis secondary to β agonist administration may be a common scenario which can be easily misinterpreted and confuse the clinical picture. Acidosis itself results in hyperventilation which could be mistaken for failure to treat the response. This may in turn lead to inappropriate intensification of treatment.
Tumefactive multiple sclerosis (MS) is a rare variant of MS. This form of MS can masquerade as neoplasm or infectious etiology. Understanding of the disease is limited to case report but it is associated with high morbidity and mortality.
Case report
A 44 year old man presented with a 2-month history of progressive right upper extremity weakness, confusion and visual change. Physical exam revealed weakness, hyperreflexia on the right side and right homonymous hemianopia. MRI of the brain showed multiple ring-enhancing lesions located in both cerebral hemispheres. CSF analysis disclosed elevated protein with positive oligoclonal bands and myelin basic protein. Stains and cultures for bacteria and mycobacteria were negative. Serologies including HIV, Toxoplasmosis, and Lyme were all negative. Patient was treated with high-dose IV corticosteroid and clinically improved. One month later, he presented with increasing confusion, aphasia and progressive weakness. Repeat MRI of the brain revealed worsening multiple ring-enhancing lesions with surrounding vasogenic edema in most lesions. High-dose corticosteroid was promptly started. There was also concern about infection, especially brain abscess; hence, intravenous ceftriaxone, vancomycin, and metronidazole were empirically given. Due to uncertainty of diagnosis, first brain biopsy at right frontal lobe lesion yielded non-specific gliosis. Repeat MRI brain showed increasing number of ring-enhancing lesions in both cerebral hemispheres. As a result, a second brain biopsy was performed, which showed an active demyelinating process consistent with multiple sclerosis. Patient experienced severe disability and was discharged to long-term facility with slowly tapered schedule of corticosteroid. He was readmitted several times and eventually family decided hospice care.
Discussion
Multiple sclerosis is diagnosed by demonstrating clinical and/or radiographic evidence of dissemination of disease in time and space1. Tumefactive MS is a term used when the clinical presentation and/or MRI findings are indistinguishable from a brain tumor2. Not all case of tumefactive MS are fulminant. Marburg variant MS is an acute rare variant of MS which has a rapidly progressive course with frequent, severe relapses leading to death or severe disability within weeks to months3. The tumefactive demyelinating lesions are defined as large (>2 cm.) white matter lesions with little mass-like effect or vasogenic edema, and post-gadolinium magnetic resonance imaging (MRI) typically showing an incomplete ring enhancement2,4. The clinical and imaging characteristics of these demyelinating lesions may mimic primary and secondary brain tumors, brain abscess, tuberculoma, and other inflammatory disorders e.g. sarcoidosis, primary sjogren’s syndrome5. As a result, tumefactive MS is frequently misdiagnosed. There are some MRI characteristics that are more suggestive of tumefactive demyelinating lesions than of other etiologies. These include incomplete ring enhancement, mixed T2-weighted iso-and hyperintensity of enhanced regions, absence of a mass effect and absence of cortical involvement2,6. Differential diagnosis of rapidly progressive neurological deficit with ring-enhancing lesions include brain abscess, primary brain neoplasm or brain metastasis, acute disseminated encephalomyelitis (ADEM) and tumefactive multiple sclerosis. Careful clinical history, CSF study, serial MRI evaluation and follow-up are usually sufficient to make a diagnosis. Some cases pose considerable diagnostic difficulty owing to clinical and radiographical resemblance to brain tumor, for which biopsy may be warranted. Pathologically, the lesions are characterized by massive macrophage infiltration, acute axonal injury, and necrosis. No specific histological features distinguished specimens derived from patients developing classic multiple sclerosis from those who had tumefactive form7. A limited number of cases of Marburg’s variant MS have been reported in the literature whereby most patients died within a period of weeks to months. Only two cases survived after one year7,8. There is no current standard treatment for this condition. Plasma exchange and Mitoxantrone are reportedly showed some promising options9,10.
Figure A: FLAIR imaging at first presentation showed lesion in both hemisphere. Figure B: FLAIR imaging at one month later showed progression of multiple lesion in both hemisphere. Figure C: T1 Post contrast imaging showed intense ring enhancement pattern in almost all lesions with mild edema and minimal mass effect. Figure D: Showed lesion view as sagittal section.
Our patient presented somewhat like a stroke with visual field defect and right hemiparesis which is unusual in MS, but MRI and CSF exam yielded a diagnosis of probable MS. Because of his abrupt clinical deterioration and impressive worsening of his MRI, concern was raised about possibility of infection or neoplasm. Hence, he received two brain biopsies, the second of which showed active demyelination, confirming the diagnosis of severe tumefactive multiple sclerosis and can be consider as a Marburg variant multiple sclerosis.
Conclusion
Marburg variant multiple sclerosis carries a high morbidity and mortality. This disease notoriously mimics other conditions leading to delay diagnosis and treatment. Absence of definitive diagnosis test apart from brain biopsy makes diagnosis, prognosis and treatment decisions difficult.
Hypertension is common but, with early detection and treatment, it is rare to see malignant hypertension. We report a patient who presented with signs suggestive of thrombotic thrombocytopenic purpura and severe hypertension, which resolved with the treatment of hypertension.
CASE REPORT:
A 34 year old African American male presented to the emergency department (ED) having experienced nausea, vomiting and diarrhoea for two days. He denied haematochezia, meleana or sick contacts at home. He complained of blurred vision without photophobia, headache and mild chest discomfort. His past medical history was unremarkable. The patient did not have any significant family history. Smoking history was significant for a pack of cigarettes daily for seven years. He reported occasional alcohol intake, and denied use of recreational drugs. On presentation, this patient’s blood pressure was 201/151 mmHg, with a mean of 168 mmHg. Pulse 103 beats per minute, respirations 20 per minute and temperature 98.4F. Physical examination was otherwise unremarkable, including absence of focal neurological deficits.
Blood tests showed: Haemoglobin 12.6 g/dl, White cell count 13.9 g/dl, Platelets 67000, Sodium 136, Potassium 3.4, BUN 24, Creatinine 2.56 and LDH 556. Chest x-ray showed cardiomegaly. A non-contrast computed tomography scan of the brain did not show any sign of stroke (haemorrhage). Urinalysis was positive for proteins 4+, a large amount of blood, 0-2 white blood cells/high power field (HPF) and 0-2 red blood cells/HPF.
Figure 1
The patient’s initial treatment whilst in the ER consisted of a Labetalol drip. His mean arterial pressure decreased to approximately 115 mmHg during the first hour, and his chest pain and headache improved with the control of elevated mean arterial pressure. Furthermore, over the next 24 - 48 hours, the patient’s blood pressure was brought down to 138/86 mmHg and his blurred vision improved significantly. Subsequently, intravenous medications were switched to an oral regimen. Blood peripheral smear from the day of admission was significant for the schistocytes (Figure 1) suggesting ongoing haemolysis. Renal ultrasound was unremarkable. His cardiac ultrasound revealed an enlarged left ventricle, however no valvular abnormality was seen. Serum calcium and thyroid stimulating hormone levels were normal, as were urine catecholamines and vanillylmandelic acid level. On two week follow up in the outpatient clinic, the patient’s platelet count and creatinine had returned back to baseline and peripheral smear did not reveal any schistocytes as the blood pressure came under better control. [Table 1]
Variable
On day 1
Day 3
Day 5
Day 6
Follow-up in 2 weeks
Haemoglobin
12.6
9.3
9.3
10.3
11.4
Platelets
67, 000
65,000
90,000
125,000
204,000
Retic. count
3.9
--
--
4.3
--
Creatinine
3.06
2.86
2.69
2.4
2.3
BUN
29
27
28
27
27
LDH
556
370
333
240
--
Troponin
0.10
0.08
0.06
0.05
--
Peripheral Smear
Schistocytes
--
--
--
No Schistocytes
Table 1
DISCUSSION:
Malignant hypertension is a medical emergency with an incidence of 1% in hypertensive patients1and is more common in the African American population2. Depending on the clinical presentation, it must be differentiated from thrombotic thrombocytopenic purpura (TTP), disseminated intravascular coagulation (DIC), glomerulonephritis and vasculitis.
Suspicion for TTP was initially high in this patient because of haemolysis, thrombocytopaenia, central nervous system (CNS) manifestations and renal insufficiency. However, TTP did not explain the presence of elevated blood pressure3,4nor the improvement in symptoms and signs with the management of this, which clearly supports our diagnosis. Rapidly progressive glomerulonephritis did not explain the CNS symptoms, and a normal prothrombin time and activated partial thromboplastin time ruled against disseminated intravascular coagulation5. The patient did not have a history of preceding diarrhoea6, which could possibly direct towards haemolytic uraemic syndrome (HUS)4. There was no history of prosthetic valves, nor clinical evidence of vasculitis. The patient’s symptoms of severe hypertension, haemolysis, thrombocytopaenia and renal failure were consistent with malignant hypertension, and treating the hypertension7gradually resolved the thrombocytopaenia, haemolysis and renal failure8.
CONCLUSION:
This case report highlights that malignant hypertension is a medical emergency which can present with features resembling a wide variety of diseases, including TTP and HUS. Using appropriate management to control the elevation in blood pressure can help reveal the underlying diagnosis.
Infection of a prosthetic total knee joint is a serious complication1 and should be diagnosed promptly2 and treated aggressively. We present an interesting case of MRSA infection of a primary total knee replacement following an IV cannula infection leading to bacteremia and subsequent infection of the knee prosthesis, complicated by stevens- Johnson syndrome .
There were many challenging issue which are outlined including diagnosis and management.
Case Report
A 63-year- old lady had an elective total knee arthroplasty for severe osteoarthritis of the knee. She had a background history of well-controlled type 2 diabetes mellitus and was on warfarin for a previous pulmanory embolism. As per the hospital protocol her warfarin was stopped before surgery until her INR was <1.5 and she was heparinised with a view of
warfarinizing after the surgery. She had an uneventful knee arthoplasty, but unfortunately one of her IV cannula site became cellulitic. She was empirically started on oral flucloxacillin after taking blood cultures and sending the cannula tip for microscopic culture and sensitivity (which is routinely done has hospital protocol for infected cannula sites).
Surprisingly the tip grew MRSA and also had MRSA bacteraemia. She became systemically unwell and septic, and was treated aggressively with parentral vancomycin for MRSA bacteraemia. She had a transeosphageal echocardiogram to rule out cardiac vegetation. She gradually improved but developed typical papular rashes over her palm, dorsum of hand, extensor surface of arm and forearm and trunk and buccal mucosa (Fig 1 and 2) .
Fig 1: Rash over the dorsum hands
Fig 2: Rash over the extensor aspects of forearm
She had a severe allergic reaction to vancomycin and the skin biopsy of the lesion confirmed that she had developed Stevens-Johnson syndrome. An alternative antibiotic was started following discussion with the specialist bone infection unit. She gradually improved over the next few weeks without any problem in her prosthetic replaced knee. At about 6 weeks post- operatively she developed severe pain and hot swelling of her replaced knee with decrease range of motion. Her inflammatory markers were markedly raised and the knee aspirate confirmed MRSA infection of the total knee replacement. She was referred to a specialist bone infection unit due to the complexity of the case, where she successfully underwent two- stage revision.
Discussion
Infection of a Knee replacement is a serious complication that requires significant hospital-based recourse for successful management3. The rate of infection of a primary knee replacement varies from 0.5- 12%1. Rheumatoid arthritis , previous surgery , diabetes mellitus are all associated with an increased risk of infection 4. Although there is no absolute diagnostic test for peri-prosthetic infection2 , a high index of clinical suspicion is essential. There has been a case report on MRSA cervical epidural abscess following IV cannulation 5, but to the best of our knowledge there has been no previous report of MRSA- infected knee arthroplasty following complications of IV cannulation. Stevens-Johnson syndrome involves rare but severe cutaneous adverse reactions related to a variety of medications including antibiotics6. Parenteral vancomycin is the first line treatment for MRSA bacteraemia. It is recognised that vancomycin is indicated in inducing Stevens -Johnson syndrome, mortalitiy being 30-100%7. It is vital that Stevens- Johnson syndrome is recognised early so that offending agents are stopped and supportive treatment commenced. Early dermatological consultation, skin biopsy and direct immunofluorescence7 are essential to confirm diagnosis so that effective treatment can be instituted.The diagnosis and management of this serious complication is complex and requires considerable recourse allocation by the patient, the hospital, the infectious disease specialist, and the orthopaedic surgeon1,5.
Robert Moots is Professor of Rheumatology at the University of Liverpool and Director for Research and Development at the University Hospital, Aintree. He is also a Consultant Rheumatologist at the hospital.
He graduated from St Mary’s Hospital, London University in 1985 and also worked at Harvard Medical School. He became a Consultant Rheumatologist at University Hospital Aintree in 1997 and the youngest full-time professor of Rheumatology and Head of Department in 2003.
Professor Moots has published extensively in rheumatology, winning the prestigious Michael Mason prize for rheumatology research. He advises the UK Department of Health and NICE. His research interests are inflammatory rheumatic diseases, in particular innate cellular immunity in rheumatoid arthritis, immunotherapy, new therapeutic targets and clinical trials.
How long have you been working in your speciality?
I’ve been working as a consultant in rheumatology since1997, when I returned to the UK from the USA. Of course I was a trainee in rheumatology for a few years before then.
Which aspect of your work do you find most satisfying?
Its hard to single out any one thing. The great fun of being Professor is that no two days are the same. My job varies so much from looking after patients, to teaching, running research and also communicating and sharing research findings with other clinicians and scientists throughout the world – giving me the opportunity to visit countries, where I would not normally have visited.
What achievements are you most proud of in your medical career?
Clinically, I often deal with rare rheumatic diseases, or situations where normal treatments have failed and other doctors have said there is “no more that can be done”. Each patient that I see in this situation, who then goes on to recover and have a normal happy life, gives me a great satisfaction. Academically, building up a successful research team of talented individuals in Liverpool, the first academic rheumatology unit in that city, has been a great privilege.
Which part of your job do you enjoy the least?
Trying to balance the demands of patient care with the many other calls on my time can be rather wearing. But nothing is worse than the ever expanding administration tasks and bureaucracy!
What are your views about the current status of medical training in your country and what do you think needs to change?
When I visit other countries to lecture, I always try to see how medicine runs there. I attend clinics and hospitals, see patients and learn how practice compares to the UK. I am pleased to note that the standard in the UK remains amongst the highest of all countries.
How would you encourage more medical students into entering your speciality?
Its hard to image why students and doctors could consider any specialty other than Rheumatology! Rheumatology provides the opportunity to see patients of all ages, develop a close rapport with patients as the diseases tend to be chronic and prevalent, perform cutting edge research to understand pathophysiological process underlying the diseases and access drugs that can make a revolution to lives with great outcomes.
What qualities do you think a good trainee should possess?
Be keen to learn, open, honest and bright. I also like trainees to challenge accepted wisdom – a considered critical approach is needed to move things forward and to keep us on our toes.
What is the most important advice you could offer to a new trainee?
Don’t accept non-evidence based dogma. Don’t learn bad habits. Be critical and try to improve things. Try to spend some time away from your unit and ideally out of your country – seeing how medicine works in other environments to get life and work in a better perspective.
What qualities do you think a good trainer should possess?
Good trainers should be excellent clinicians, inspirational leaders and listeners with patience. If you know someone like this, you should really treasure them!
Do you think doctors are over-regulated compared with other professions?
No – but I fear that we are getting there in the UK.
Is there any aspect of current health policies in your country that are de-professionalising doctors? If yes what shouldbe done to counter this trend?
With a recent change in government in the UK and major changes to the Health Service planned, it’s a little too early to tell. We have to be vigilant though.
Which scientific paper/publication has influenced you the most?
For much of my working life, I was focused on the T cell as the major driver for diseases such as rheumatoid arthritis. The paper that changed that was: Edwards SW, Hallett MB. Seeing the wood for the trees: the forgotten role of neutrophils in rheumatoid arthritis. Immunol Today.1997 Jul;18(7):320-4. This crucial paper from Steve Edwards, the world leader in neutrophil biology opened my eyes to a whole new field of work. I didn’t know at the time that I would eventually have the privilege of working with Steve.
What single area of medical research in your specialty should be given priority?
That’s an easy one – it should be whatever my group are working on at the time.(I just wish that were the case!)
What is the most challenging area in your specialty that needs further development?
Many rheumatic diseases such as rheumatoid arthritis can be treated extremely successfully (with patients enjoying a full remission) if they can access the right drugs at the right time. There is still much variability in time to diagnosis and in provision of appropriate medications – the challenge is to ensure that best practice can be rolled out more effectively.
Which changes would substantially improve the quality of healthcare in your country?
There needs to be a greater understanding of the importance of rheumatic diseases in the UK. These conditions are prevalent, may cause significant morbidity (and indeed mortality), cost the nation considerably in reduced productivity and in disability payments – yet many of these conditions can be treated most effectively.
Do you think doctors can make a valuable contribution to healthcare management? If so how?
Its crucial that doctors are fully engaged in management. We are in the best position to be advocates for our patients but cannot do this effectively without understanding the health care system and take the lead in ensuring this works for the best.
How has the political environment affected your work?
The consequences of the recent change in Government in the UK are likely to be considerable for the National Health Service. This will involve major changes to the work of staff at all levels. It is too early to know the full extent of this – but we all wait with trepidation
What are your interests outside of work?
With so much to do, its hard to find the time for much else apart from relaxing with my family. I travel a lot and especially enjoy taking my children with me. My 10 year old has heard me lecture so much that I suspect she can give my talk for me (and do it better). She has also taken to asking questions at the end of my lecture, which always scares the chairperson of the meeting!
If you were not a doctor, what would you do?
I’m not sure that I would be fit for anything else!
A 73 year old lady presented for assessment of her recurrent right sided pleural effusion. She had a history of gallstones and underwent open cholecystectomy. One month after surgery the patient had recurrent pleural effusion requiring thoracocentesis on a monthly basis. On the chest x-ray, the pleural effusion was seen exclusively on the right side occupying the whole right hemithorax.
The pleural fluid was transudative on multiple occasions and there was no evidence of malignant cells. Her echocardiography revealed preserved cardiac function. An abdominal ultrasound showed findings of cirrhosis and splenomegaly consistent with portal hypertension.
Image 1
Computerised tomography (CT) of the chest and abdomen revealed a large right-sided pleural effusion and minimal ascites (Image 1). An ultrasound guided paracentesis was performed with difficulty and only 17cc of fluid.was obtained. The abdominal fluid showed similar consistency as the pleural fluid. The blood workup at the same time was unremarkable.
Image 2
Intra-peritoneal administration of 99mTc-sulphur colloid was attempted but failed in the absence of ascites. Computed tomography with three dimensional reconstruction at the diaphragmatic level revealed a defect in the posterior aspect of the right hemidiaphragm (Image 2 black arrow) and also revealed irregular contours of the liver, an indirect sign of diaphragmatic defect (Image 2 white arrow).
The patient declined any surgical intervention at that point including the option of pleurodesis. She was started on diuretics and a low salt diet with significant improvement.
Discussion:
Pleural effusion due to hepatic cirrhosis and ascites is well known, but hepatic hydrothorax in the absence of ascites is a rare complication. We report a case of liver cirrhosis with a large and recurring right sided pleural effusion that had an apparent abdominal source in the absence of ascites. We review the characteristics and treatment for hepatic hydrothorax in the absence of ascites.
Hepatic hydrothorax is defined as the presence of significant pleural effusion in a cirrhotic patient without primary pulmonary or cardiac disease1. Postulated mechanisms for the development of pleural effusions in patients with hepatic cirrhosis include: hypoalbuminemia and decreased oncotic pressure leakage of the plasma from the hypertensive azygos vein, lymphatic leak from the thoracic duct, passage of ascitic fluid to the pleural space by way of lymphatic channels in the diaphragm, and transfer of peritoneal fluid directly via diaphragmatic defects2.
The usual unilaterality of hepatic hydrothorax could be attributed to a congenital factor, but not to physiologic mechanisms3. The most likely explanation appears to be that ascitic fluid passes through congenital or acquired fenestrations in the diaphragm directly into the pleural space2. The description of hepatic hydrothorax in the absence of ascites is very rare1. The flow of the ascitic fluid into the pleural space equaled the rate of ascites production in patients with this entity3.
The composition of pleural fluid from hepatic hydrothorax is similar to that of ascitic fluid. Pleural effusions associated with portal hypertension are always transudative1. Nuclear scans can be performed to establish the diagnosis of hepatic hydrothorax with fairly high accuracy. Intra-peritoneal administration of 99mTc-human serum albumin or 99mTc-sulphur colloid can be used to demonstrate the communication between the peritoneal and pleural space. Recent advances in radiological imaging have enabled investigators to examine in detail the diaphragmatic defects responsible for the development of hepatic hydrothorax1.
The management is challenging and frequently associated with poor outcomes in most cases. Dietary restriction of sodium intake and the addition of diuretics is the initial approach. Thoracocentesis can be performed in patients with dyspnoea due to hepatic hydrothorax for immediate relief of symptoms. When thoracocentesis is required too frequently in patients on maximal sodium restriction and optimal diuretics, alternative treatment options must be considered1, 3.
Over the last few years, new insights into the pathogenesis of this entity have lead to improved treatment modalities such as portosystemic shunts (TIPS) and video-assisted thoracoscopy (VATS) for closure of diaphragmatic defects. Both, though temporary measures, are perhaps the best available bridging to liver transplantation in selected patients with refractory hepatic hydrothorax2, 3.





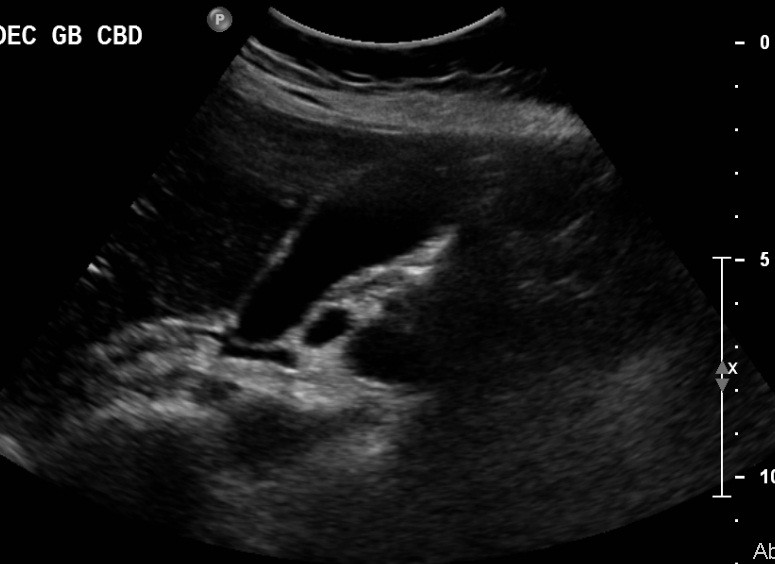
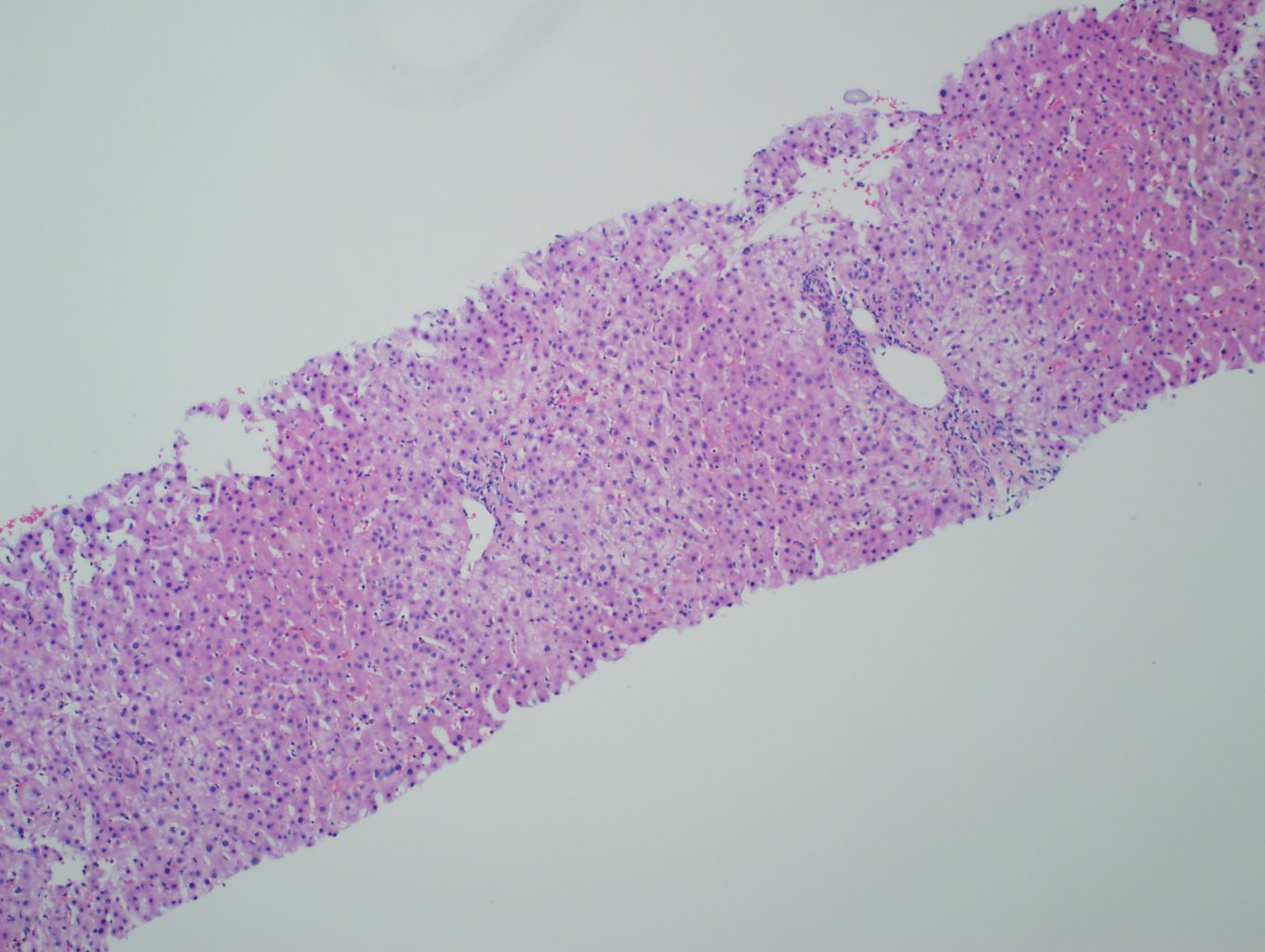
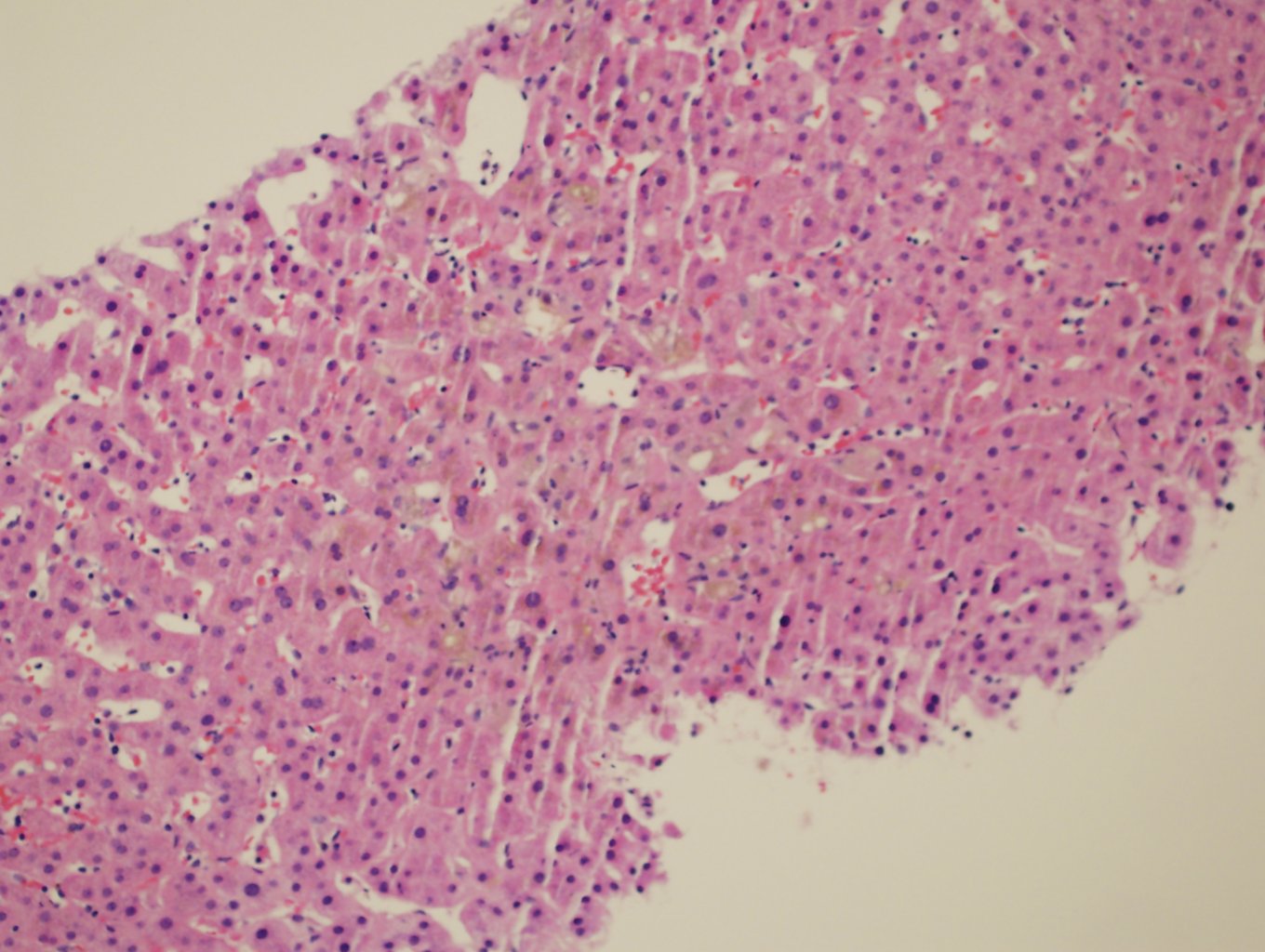
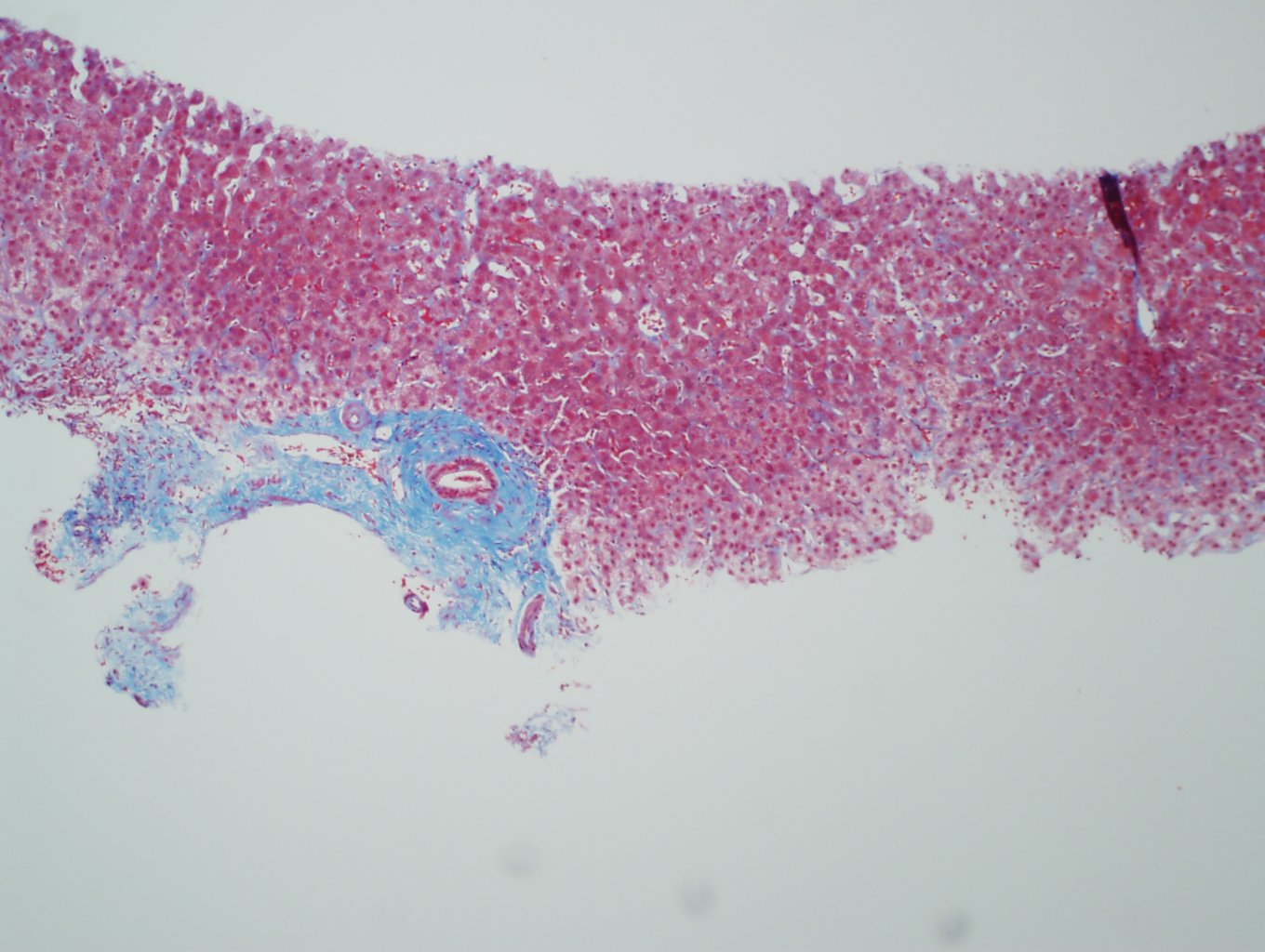
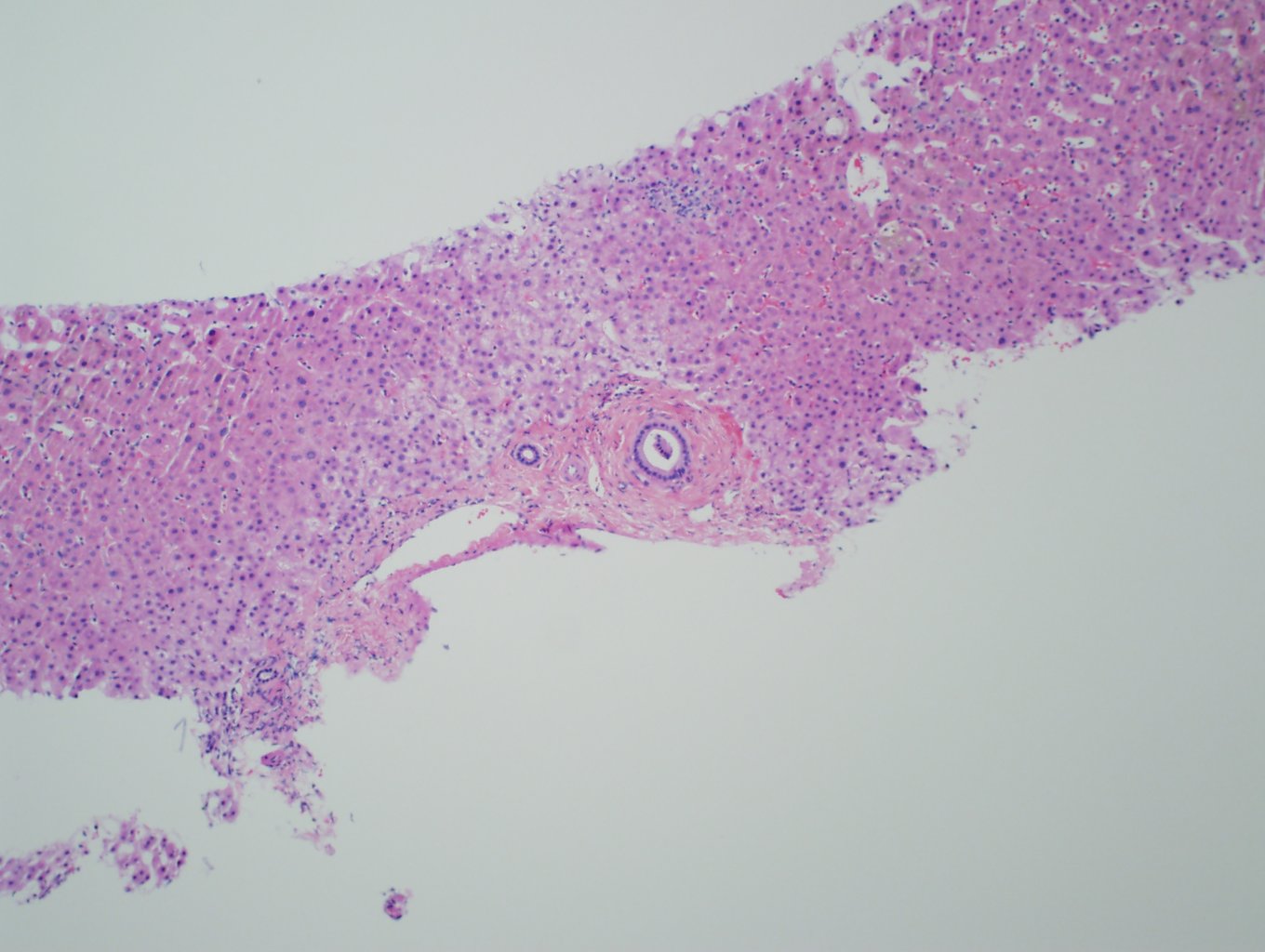
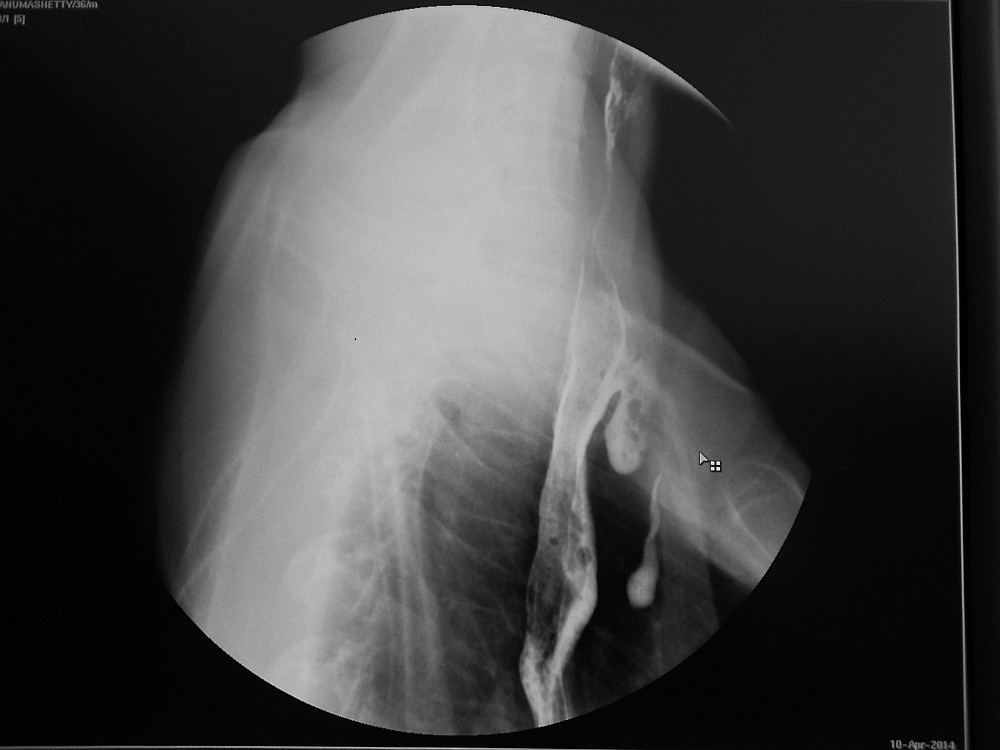
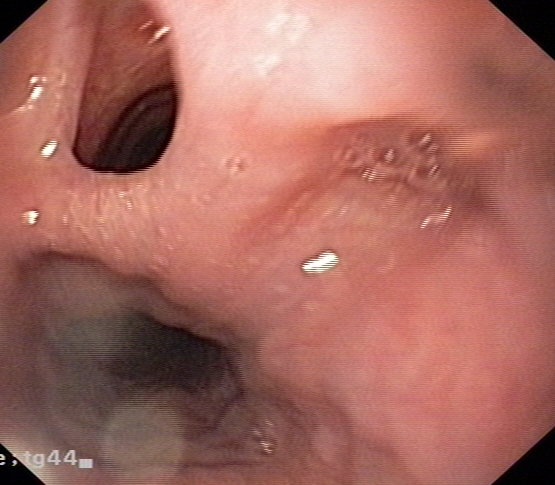
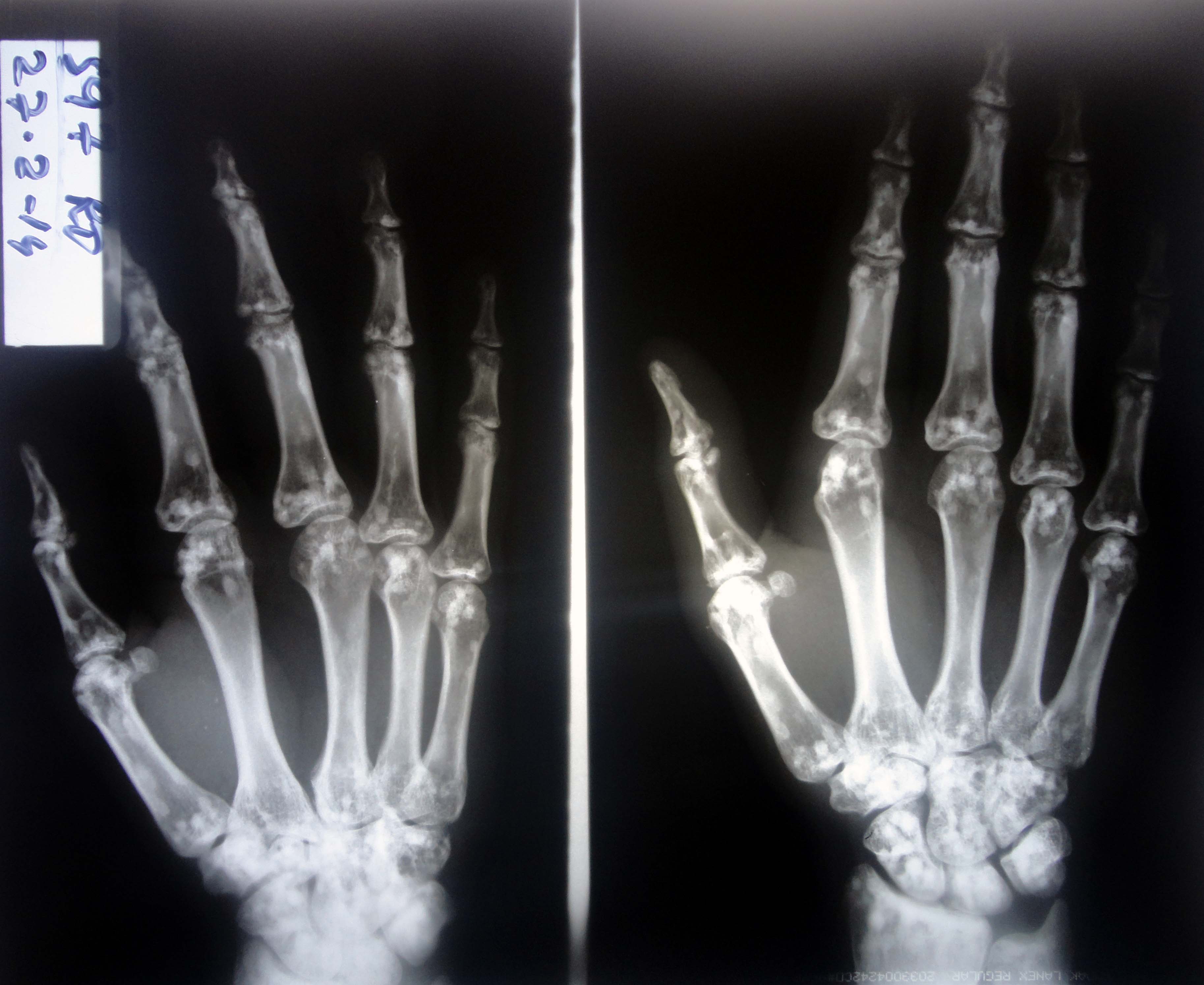
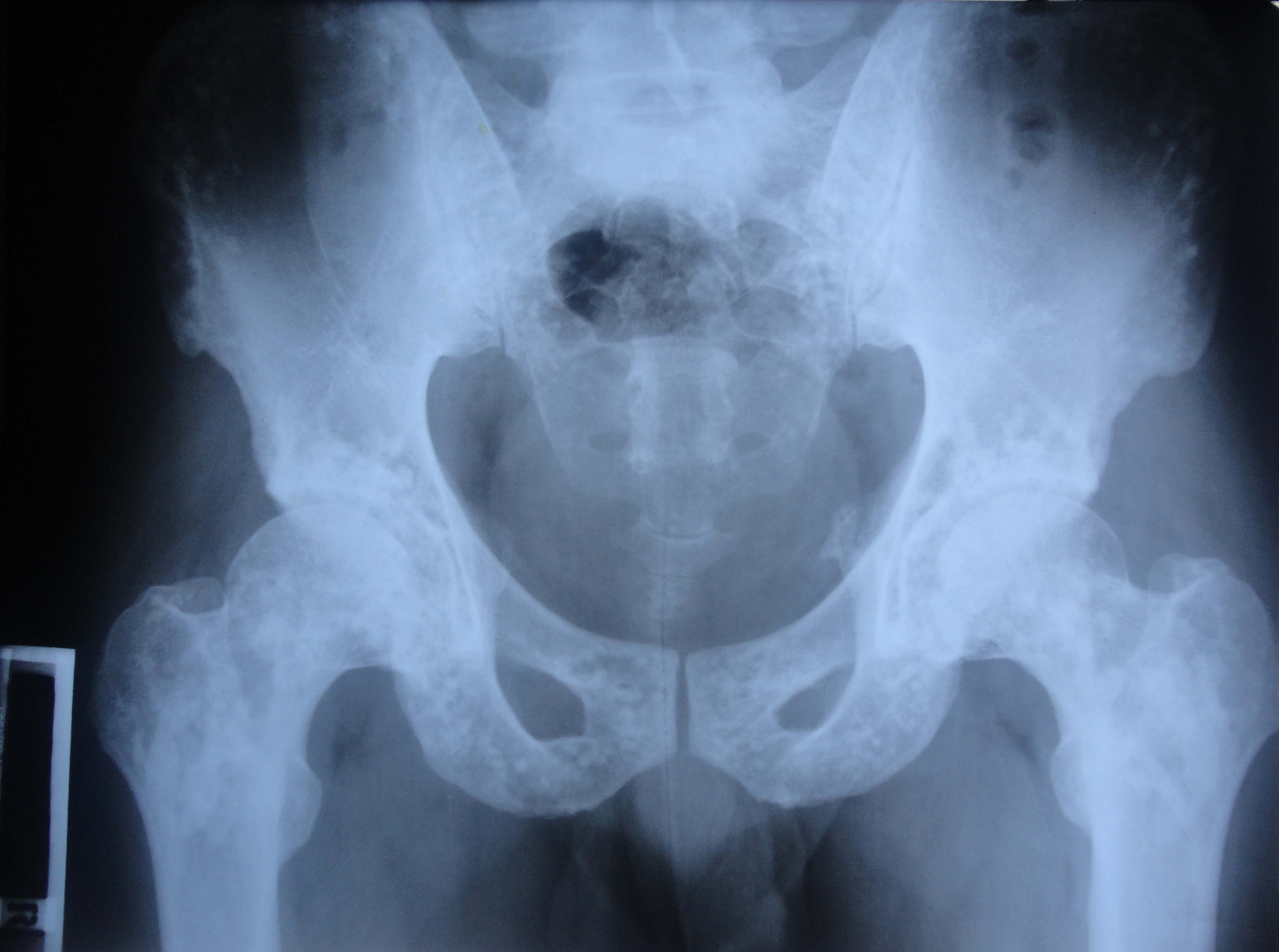
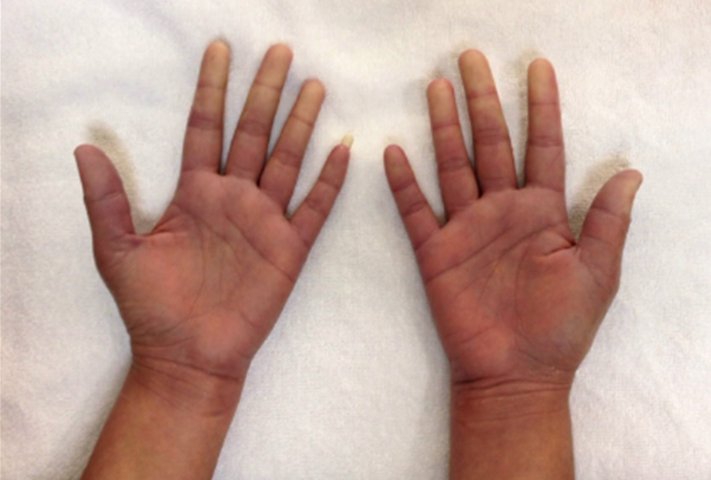
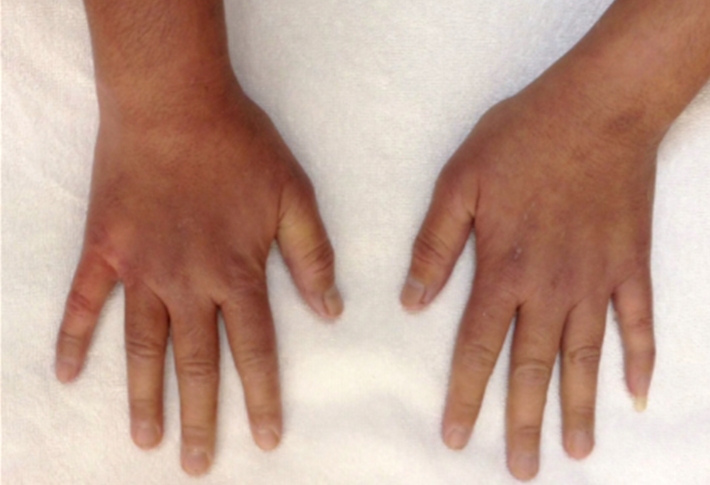
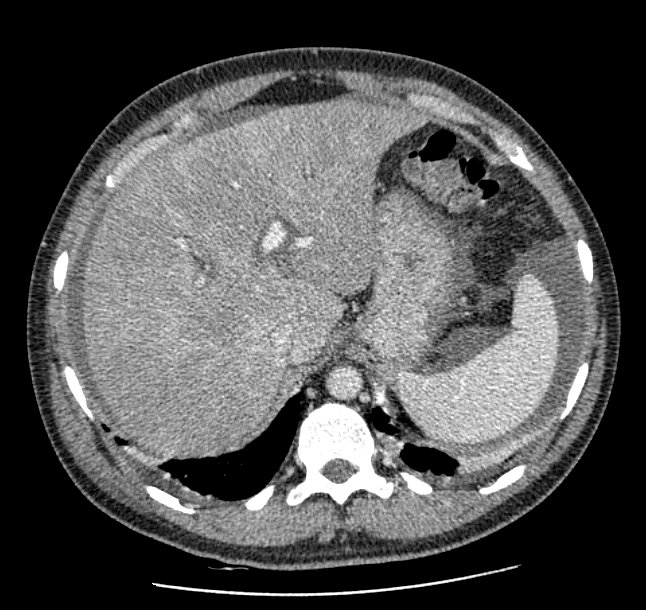
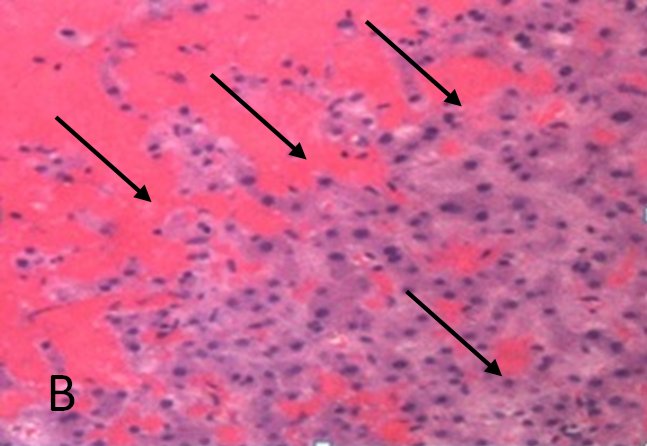
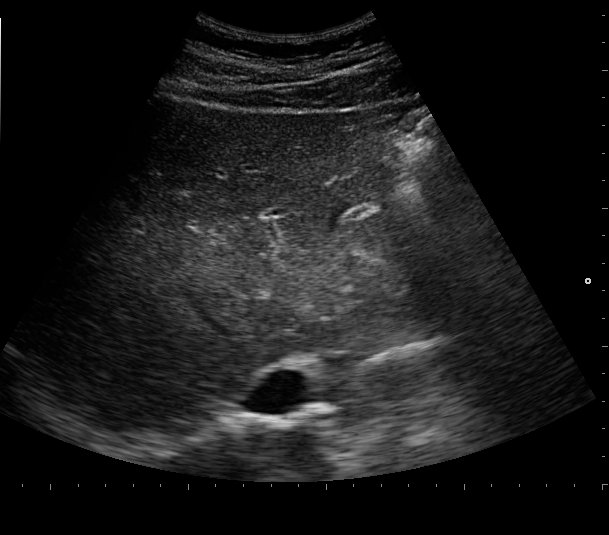
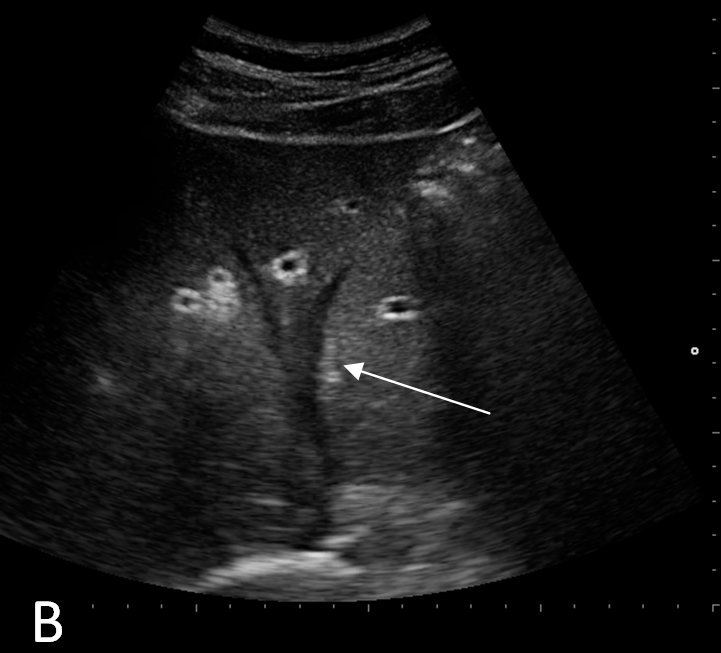
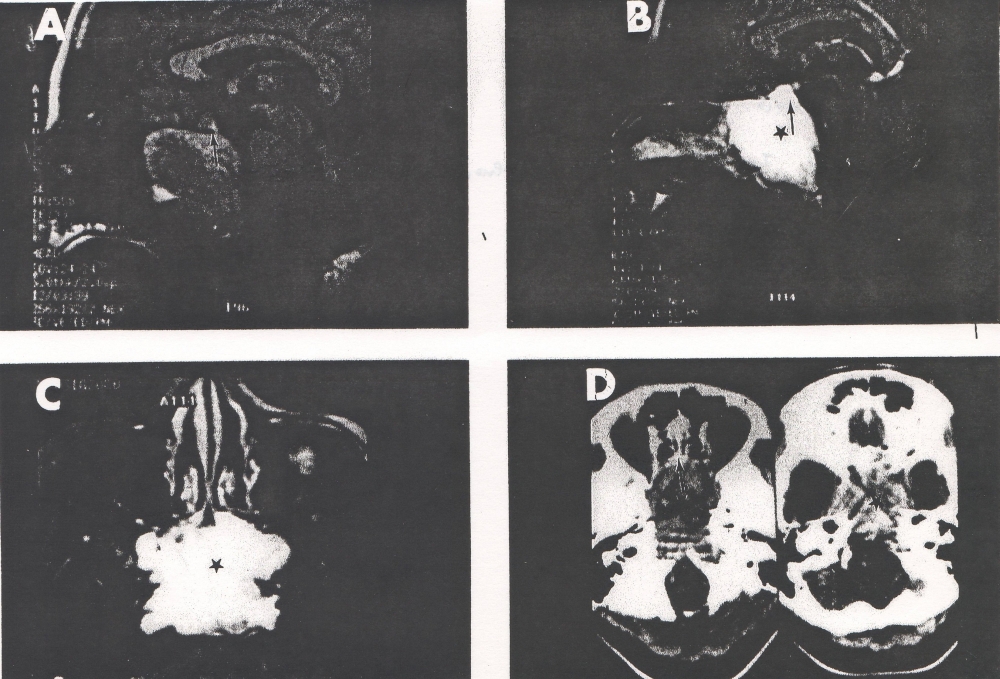
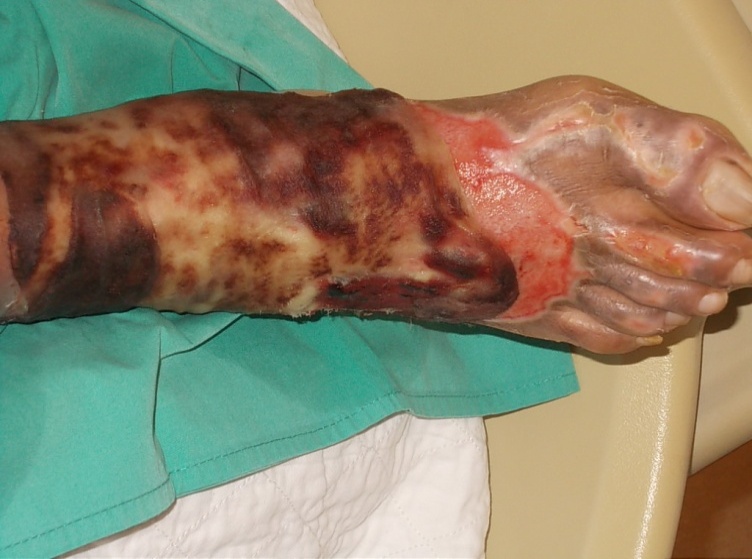
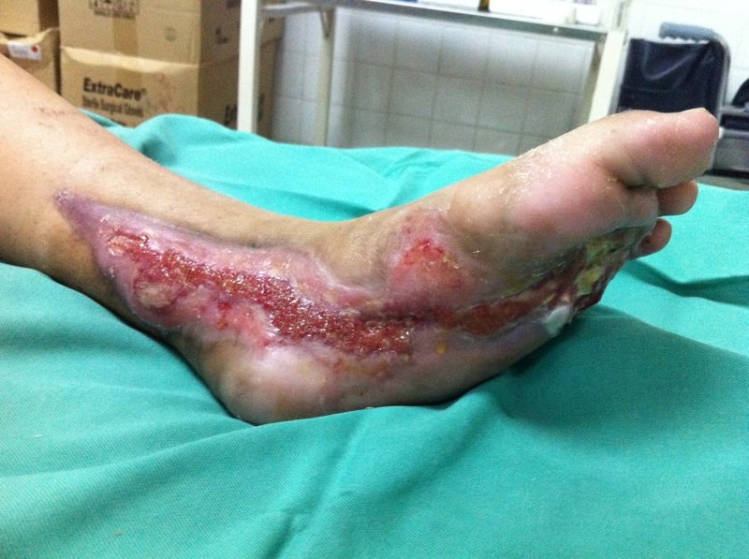
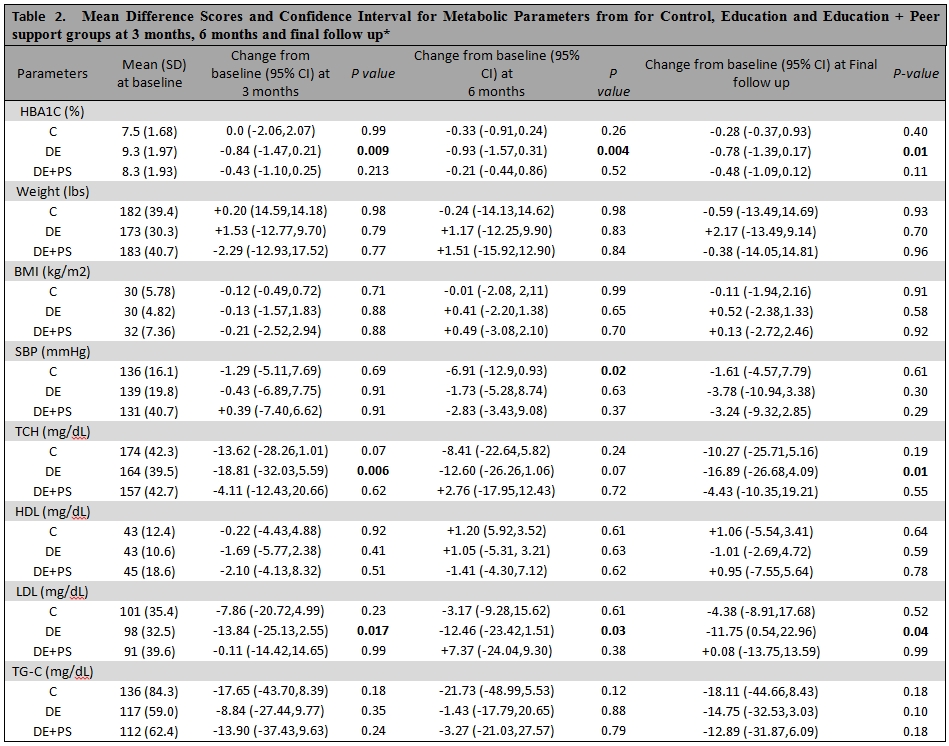

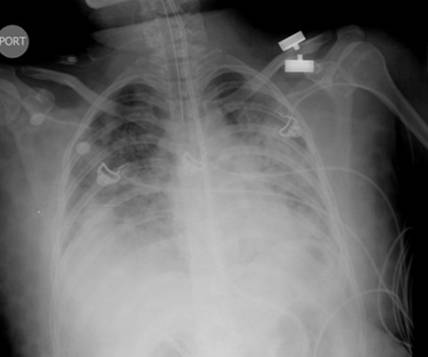
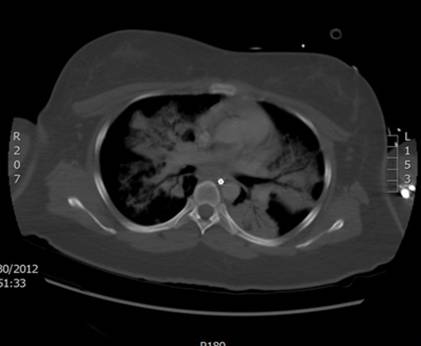
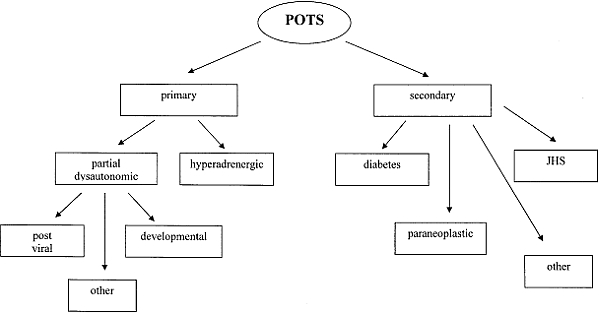
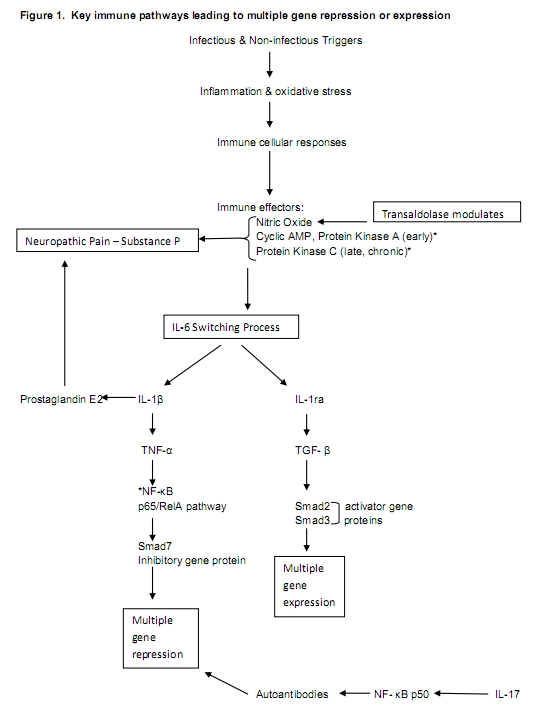
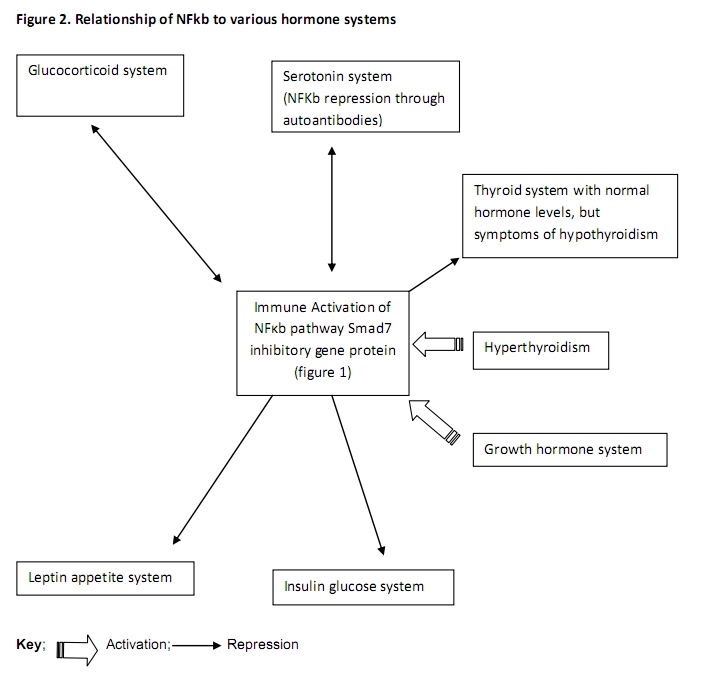
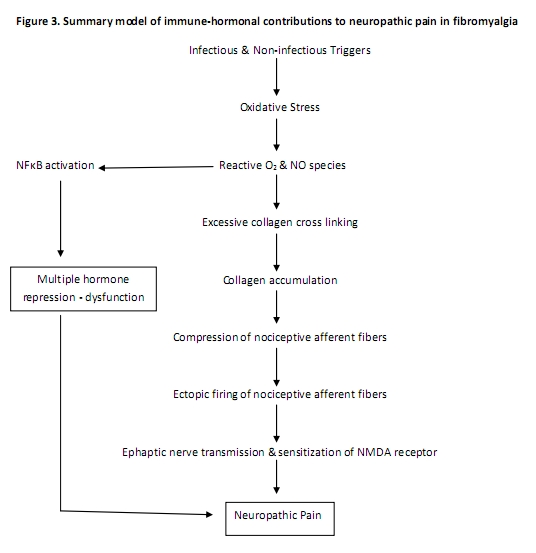
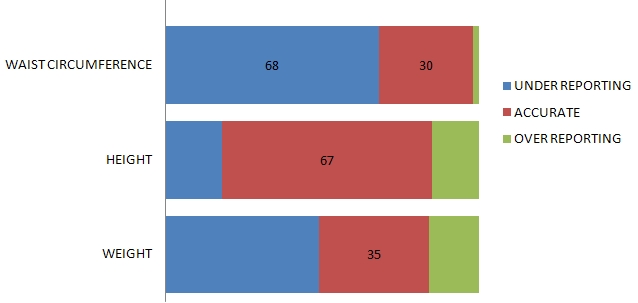
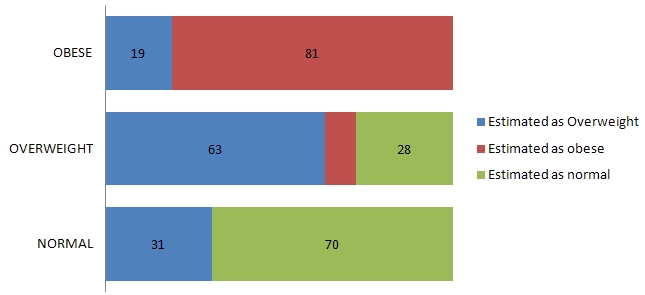
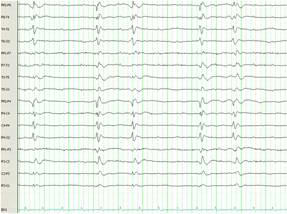
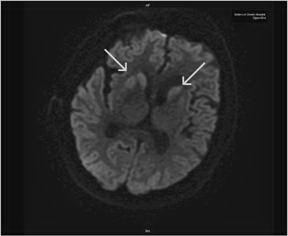
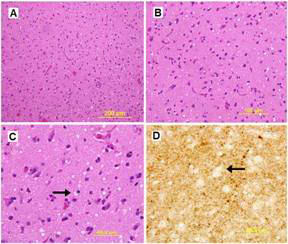
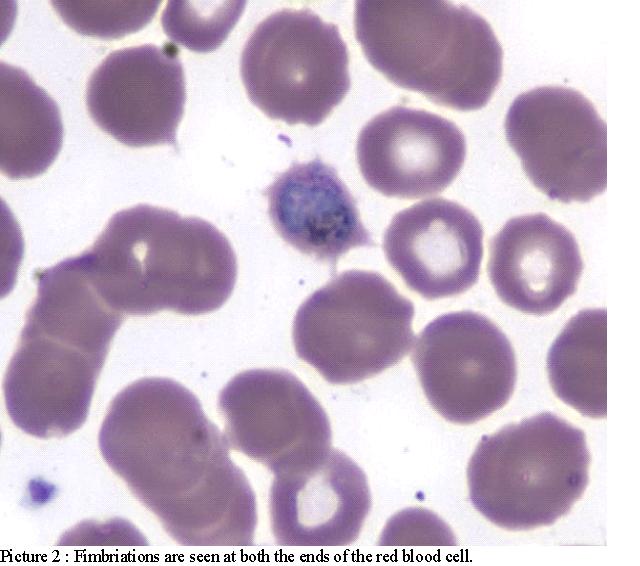

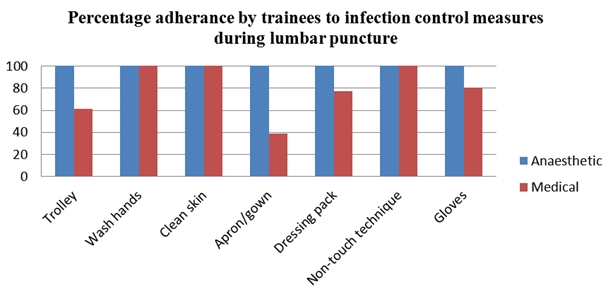
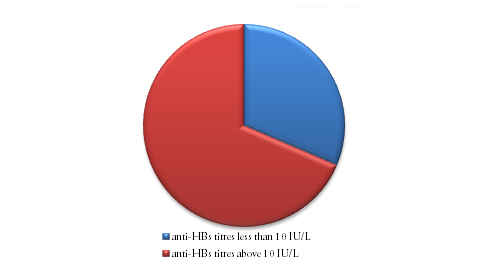
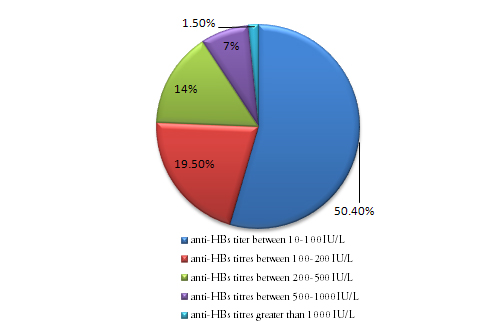
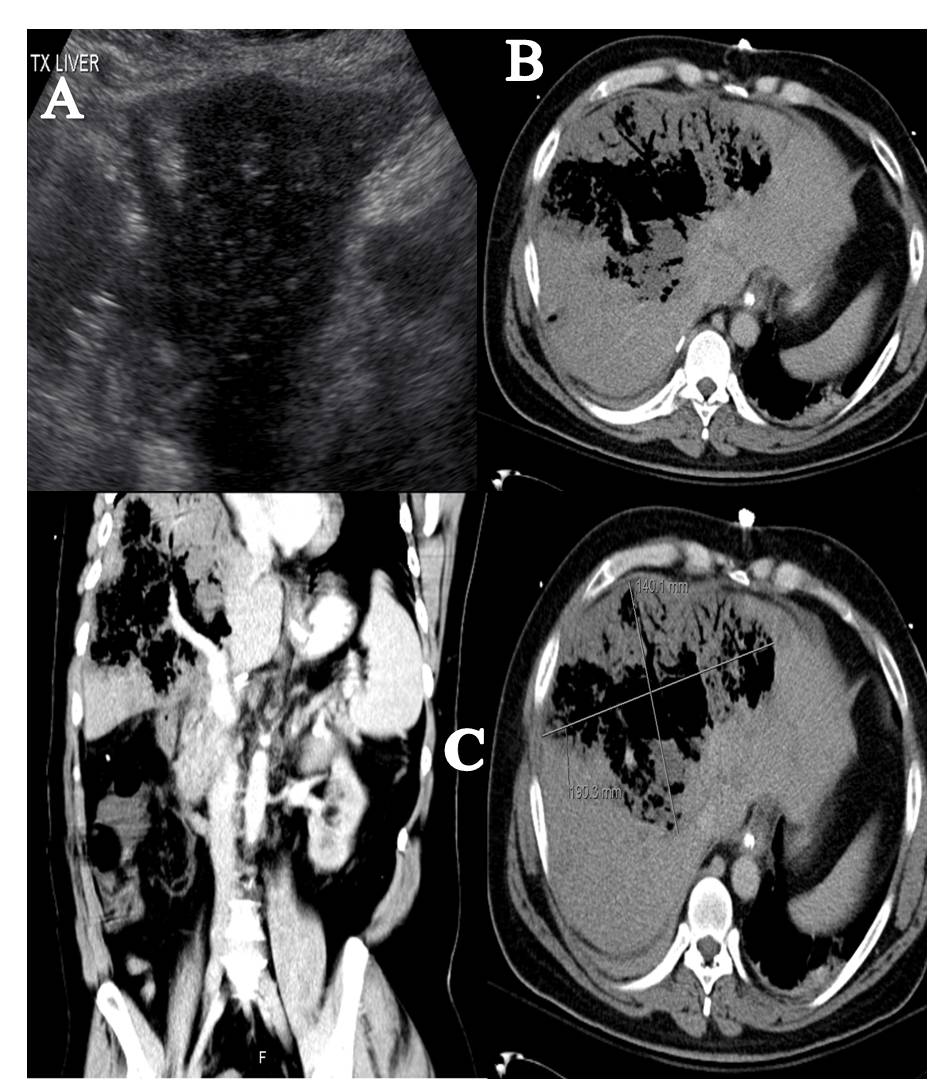
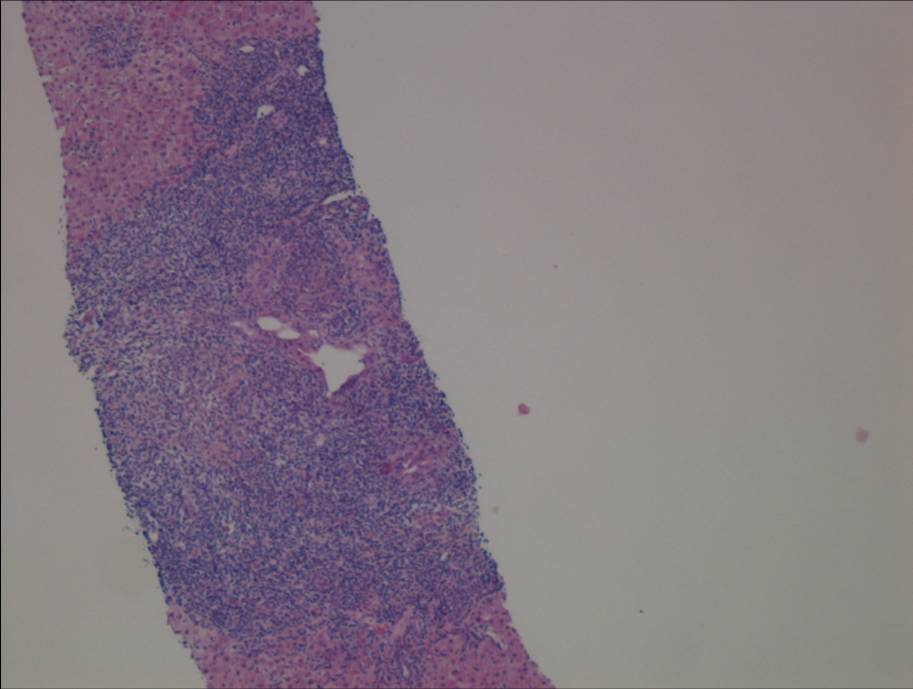
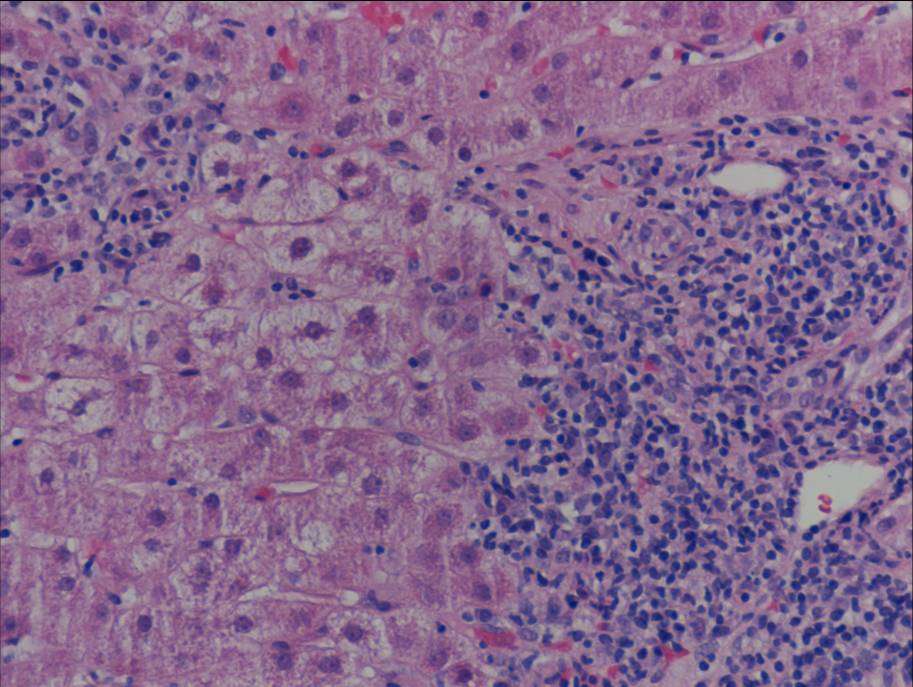
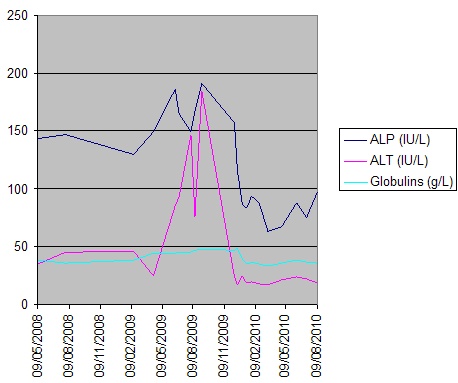

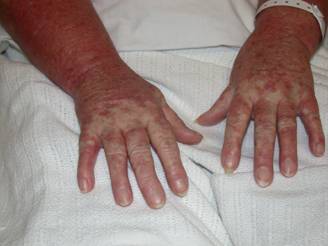
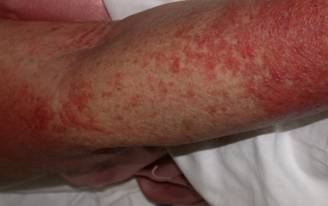

.jpg)
