Various classes of medications have been known to cause drug induced liver injury (DILI), however not much literature has been published regarding angiotensin converting enzyme inhibitors (ACE-I) causing DILI. Recent years have seen tremendous increases in ACE-I prescriptions for coronary artery disease, diabetic nephropathy and hypertension. We report the first case of lisinopril induced hepatitis via a cholestatic mechanism.
Case:
A 47 year old female with history of diabetes mellitus type 2, hypertension, chronic kidney disease (CKD)stage III, non-obstructive coronary artery disease was admitted with complains of generalized weakness, lack of appetite, yellow discoloration of skin and eyes, dark urine and white stools for 1 week prior to admission. She denied history of alcohol abuse, past liver disease, illicit drug use, recent sick contacts, fever, chills, travel. Current patient medications included lisinopril, pioglitazone, furosemide, atenolol, metformin and detemir. Patient was started on these medications about 2 years prior to admission. Patient received enalapril for 5 months before switching to lisinopril about 2 years prior to presentation.
Physical examination was positive for icteric sclera, icteric skin; negative for spider nevi, palmar erythema and asterixis. Exam did not reveal hepatomegaly or splenomegaly. Labs showed hemoglobin 8.7 gm/dl, normal white count and platelet, normal C-reactive protein, alkaline phosphatase (ALP) 750 U/L, aspartate transaminase (AST) 169 U/L, alanine transaminase (ALT) 210 U/L, gamma-glutamyl transferase (GGT) 813 U/L, total bilirubin 13.4mg/dl with conjugated fraction 7.7mg/dl, ammonia level 64. Prior to initiation of lisinopril ALP was 87 U/L, GGT 53 U/L, with AST18 U/L, ALT 11 U/L and normal bilirubin fractions. Hepatitis A, B, C and D serologies were negative. Serum acetaminophen level was normal. Anti nuclear antibody (ANA), anti- mitochondrial antibody (AMA), anti-endomysial antibody, c-anti-neutrophil cytoplasmic antibody (ANCA), p-ANCA was negative. Anti smooth muscle antibody was weakly positive in titre of 1: 40. Creatine kinase, ceruloplasmin and alpha -1 antitrypsin level were normal. Quantiferon gold was negative. Lipid panel was deranged with cholesterol level 1017 and low density lipoprotein 1006, triglycerides 255.
Ultrasonography and magnetic resonance imaging abdomen showed hepatomegaly 17.5cms but was negative for fatty infiltration of liver, stones, cirrhotic features or dilation of biliary tree. Liver biopsy was done which showed mild portal chronic hepatitis with lymphocytic infiltration (Fig: 1), cholestasis (Fig: 2), mild portal fibrosis (Fig: 3), negative for bile duct damage (Fig: 4), negative for cytoplasmic inclusion. Congo red stain was negative for amyloid.
Figure 1: Mild hepatitis with portal tract lymphocytic infiltration.
Patient was treated with fluids, anti-histaminic, ursodeoxycholic acid. Patient was unable to tolerate coleveselam. Impression was drug induced hepatitis, lisinopril was discontinued and patient improved clinically and biochemically. Discharge labs two weeks after discontinuation of lisinopril showed AST 80 U/L, ALT 70 U/L, ALP 1045 U/L and GGT 1212 U/L; total bilirubin of 3.93 mg/dl with conjugated fraction 2.43mg/dl. Patient was discharged uneventfully with follow up in Hepatology clinic. Six months after discontinuation of lisinopril ALP was 199 U/L, GGT 168 U/L with AST 19 U/L, ALT 17 U/L, total bilirubin 0.9mg/dl and conjugated bilirubin 0.21mg/dl. Patient is currently asymptomatic and icterus has resolved.
Discussion:
ACE-I has been used widely for coronary artery disease, hypertension and diabetic nephropathy and approximately 159 million prescriptions for ACE-I are written annually. Recent JNCC guidelines recommended ACE-I to be used as first line anti-hypertensives for patients with CKD and diabetes. The common side effects known about ACE-I use are cough and angioedema, hypersensitivity. However not much awareness exists regarding ACE-I induced hepatotoxicity. It is important to consider ACE-I as an etiology for drug-induced liver injury (DILI) since continuation of the ACE-I beyond onset of hepatitis is fatal1.
Literature review shows multiple reports of DILI with captopril2, 3, ramipril4, fosinopril5, 6 and enalapril.2,7 Most commonly implicated ACE-I are enalapril and captopril. The usual presentation for ACE-I induced hepatotoxicity is cholestasis mediated hepatitis. Till date there have been four case reports published reporting lisinopril as cause of hepatitis 1, 8, 9 All 4 cases of lisinopril induced hepatotoxicity have shown a hepatocellular pattern of liver injury and did not show any cholestatic features. We report the first case of lisinopril induced cholestasis mediated hepatotoxicity.
In our case, patient had received enalapril for 5 months before initiation of lisinopril; however patient developed symptoms 2 years after initiation of lisinopril. The patient had no past medical history of liver or biliary tract disease. A thorough investigative workup was negative for autoimmune and other viral causes of hepatitis. Older case reports of lisinopril induced toxicity have shown similar histopathological findings of portal inflammation by lymphocytes without centrilobular zonal necrosis.9 There are various theories regarding possible mechanisms for DILI with lisinopril, namely terminal proline ring mediated bile stasis8, 10 and hypersensitivity to the sulfhydryl group.2 Discontinuation of metformin, pioglitazone, furosemide, atenolol and detemir did not result in clinical or biochemical improvement. Patient was initially continued on lisinopril since suspicion was low and then later discontinued. Similarity in histopathological findings along with a strong temporal relationship between lisinopril withdrawal and improved biochemical and clinical scenario, with absence of other constitutional symptoms and eosinophilia strongly point toward lisinopril-induced hepatotoxicity.
Our case had a long period of latency between drug intake and onset of hepatic injury which is consistent with other published reports of lisinopril induced hepatocellular injury9, 10, 11; however the mechanism responsible for latency or hepatotoxicity remains unclear. Earlier report postulate metabolic idiosyncratic reaction as a possible molecular mechanism for hepatocellular injury9. However our case is unique as the primary mode of injury appears to be cholestatic. Since our patient received enalapril before initiation of lisinopril without any adverse events, this case adds further controversy as to whether this patient could have been safely continued on other ACE-I except lisinopril or whether she would have developed hepatotoxicity if enalapril was continued. This case highlights further need for research to evaluate ACE-I induced hepatotoxicity. Currently the awareness for ACE-I induced liver injury is low and there are no guidelines guiding physician to monitor for possible hepatic adverse events. Further research is needed to delineate the mechanism by which ACE-I cause hepatotoxicity and to define possible risk factors.
Conclusion:
Discontinuation of ACE-I beyond recognition of DILI hepatitis usually leads to normalization of liver enzymes, however continuing or reinitiating ACE-I can be severe and potentially fatal. Thus, it is important to be aware of ACE-I as a possible cause of DILI, which can present with either hepatocellular or cholestatic mechanism and to promptly discontinue ACE inhibitor use. Currently there are no guidelines in place for monitoring of liver enzymes following initiation of ACE-I and more research is required to delineate possible mechanisms and prevent further DILI in such patients.
We report the case of 36 year old white Caucasian female who used to work as a driving instructor and living with her parents.
She has a diagnosis of congenital adrenal hyperplasia (21 hydroxylase deficiency) and is on long term corticosteroid replacement (prednisolone 4 mg once daily and fludrocortisone
100 mcg once daily) and she is under the care of an endocrinologist.
She was referred for psychiatric evaluation with “anxiety and depressive symptoms” and failure to respond to antidepressant treatment which was prescribed by her General Practitioner.
During the psychiatric assessment, she reported long history of recurrent episodes of elevated mood and depression dating back to her late teens with clear description of distinct periods of mood elevations lasting for few weeks and longer periods of persistent low mood. There was no history of psychotic symptoms and no family history of mental illness.
She was diagnosed with bipolar affective disorder and failed to achieve remission of symptoms on two different antipsychotic medications (quetiapine and olanzapine) and anticonvulsant medication (sodium valproate) before starting lithium carbonate.
The introduction of lithium and gradual titration resulted in significant improvement in her symptoms and mood stability. However, few months later, she reported relapse in her symptoms (mainly reporting features of bipolar depression) despite adequate lithium levels.
She agreed on the introduction of lamotrigine as an adjunctive medication to lithium. The initial dose of lamotrigine was 25 mg daily for two weeks in line with dose recommendation from manufacturer and drug guides.
On the same day of lamotrigine introduction, the patient started to experience visual hallucinations that she never had before (please see patient’s perspective for detailed description of her hallucinations).
With the dose of lamotrigine increased to 50 mg daily after the initial two weeks, she started to report worsening of these abnormal perceptions which developed into more complex visual and auditory hallucinations.
More importantly, there was no evidence of accompanying manic symptoms or severe depressive symptoms to explain these symptoms and also no alcohol or drug use.
Following a psychiatric review, the dose of lamotrigine was reduced to 25 mg which resulted in immediate reduction in the intensity of the abnormal perceptions. When the lamotrigine was eventually stopped after one week, there was complete cessation of abnormal perceptions.
Lamotrigine was never re-started again and she was maintained on a combination of lithium and quetiapine with good effect.
Investigation:
We used the Naranjo Adverse Drug Reaction Probability Scale (1) to determine the likelihood of whether an adverse drug reaction is related to this specific drug or may be related to other factors. This tool examine factors such as the temporal association of drug administration and event occurrence, alternative causes for the event, drug levels, dose – response relationships and previous patient experience with the medication.
The probability of the adverse drug reaction is concluded from the total score (Definite if the overall score is 9 or greater, Probable for a score of 5-8, Possible for 1-4 and Doubtful if the score is 0).
Questionnaire
1. Are there previous conclusive reports on this reaction? Yes (+1)
2. Did the adverse events appear after the suspected drug was given? Yes (+2)
3. Did the adverse reaction improve when the drug was discontinued or a specific antagonist was given?
Yes (+1)
4. Did the adverse reaction appear when the drug was re-administered? Do not know or not done (0)
5. Are there alternative causes that could have caused the reaction? No (+2)
6. Did the reaction reappear when a placebo was given? Do not know or not done (0)
7. Was the drug detected in any body fluid in toxic concentrations?
No (0)
8. Was the reaction more severe when the dose was increased or less severe when the dose was decreased?
Yes (+1)
9. Did the patient have a similar reaction to the same or similar drugs in any previous exposure?
No (0)
10. Was the adverse event confirmed by any objective evidence? Do not know or not done (0)
Scoring 7 (Probable Adverse drug reaction)
Discussion:
Lamotrigine is a phenyltriazine derivative used as an anticonvulsant drug with established mood stabilising properties. In the United Kingdom, it is recommended for use in bipolar affective disorder according to the guidelines from the National Institute of Health and Care Excellence (2) and the British Association for Psychopharmacology (3).
We performed a literature search to find similar case reports. We searched the following databases using the keywords (lamotrigine AND hallucinations): Complementary Medicine (AMED), British Nursing Index BNI), Cumulative Index to Nursing and Allied Health Literature (CINAHL), Excerpta Medica Database (EMBASE), Health Business Elite (HMIC), Medline, PsycINFO and Health Management Information Consortium (HMIC).
The search returned 57 results. Only 8 articles discussed hallucinations and other psychiatric symptoms as side effects associated with lamotrigine and therefore were included in this review.
Psychotic symptoms have been reported with the use of lamotrigine (both as an anticonvulsant or mood stabiliser) but this reaction is mainly seen in patients with history of epilepsy. One study reported 4.8% incidence of psychiatric and behavioural side effects with lamotrigine in 546 patients with epilepsy. (4)
Another study on paediatric patients showed that reversible visual and auditory hallucinations were reported in one patient among 9 patients with epilepsy who received lamotrigine treatment (mean age 5 years). (5)
Villari et al published a literature review on psychiatric symptoms related to lamotrigine and included case reports documenting full acute psychotic episodes hallucinations and affective switching in patients with and without history of epilepsy.(6)
They found one case report on hallucination with lamotrigine in bipolar patient without epilepsy. In patients with epilepsy, they reported two cases reports and one case series (total number of patients 9) developing psychotic symptoms following lamotrigine and one randomised controlled trial in which four out of 216 patients stopped lamotrigine due to psychotic symptoms (including hallucinations and delusions).
The authors concluded that majority of the case reports concluded that these symptoms were lamotrigine-induced due to the temporal association with lamotrigine treatment and favourable outcome following drug withdrawal. It also appeared that more case reports were from patients with epilepsy, suggesting lower incidence in patients without this condition.
Chistyakova and Amos (7) reported a case of delirium associated with lamotrigine use. The dose of lamotrigine was increased from 200 to 400 mg over two weeks prior to her admission. The patient reported visual and auditory hallucination with confusion. She took an accidental overdose of her medication (200 mg of fluoxetine and 2800 mg of lamotrigine) due to her confusion and medications were stopped.
The authors concluded that delirium may result from lamotrigine toxicity or drug interaction with fluoxetine.
Uher and Jones in 2006 (8) reported a case of a 42-year-old woman with bipolar affective disorder with comorbid alcohol abuse and no history of neurological illness.
The patient tolerated an initial dose of lamotrigine 50 mg/day but following a dose increase to 100 mg/day, she reported vivid dream-like experiences and subsequently she reported visual hallucinations. These symptoms subsided over a few days when the dose was decreased to 50 mg/day.
The authors suggested a causal association through this dose dependent effect but also pointed out that the concurrent alcohol abuse may have been a contributing factor.
They also highlighted the paucity of case reports documenting this rare adverse reaction and identified two similar case reports in their references (which we were unable to get their full text) and a third paper reporting hallucination in 2 out of 108 patients with epilepsy on a combination of lamotrigine and sodium valproate (9)
Hallucination with lamotrigine when combined with valproic acid was also reported in a case report by Roberts et al (10) in 14 year old girl with epilepsy when it was added to valproic acid and it was suggested that this adverse effect may be due to an interaction between the two medications causing lamotrigine half-life to triple with valproic acid.
Learning points:
Lamotrigine is an anticonvulsant with an established role in management of bipolar affective disorder, particularly for the treatment and prevention of depressive episodes.
However, it appears to be associated with variable incidence of psychiatric symptoms which should be known to the prescriber and patient.
These adverse effects are mainly seen in patients with history of epilepsy but can occur in patients with mental health problem without epilepsy.
Different mechanisms for inducing these psychiatric symptoms have been suggested, including idiosyncratic reaction, lamotrigine toxicity as a result of concomitant use of another drug that affect lamotrigine metabolism (e.g., valproic acid) and delirium.
Examples of these psychiatric symptoms including affective switches in depressed patients with bipolar disorder, hallucinations in depressed patients, delirium and psychotic symptoms (mainly hallucinations and delusions) in patients with or without epilepsy.
Reversible and severe psychiatric disturbances associated with lamotrigine therapy are rarely reported in literature and more research is needed to identify population at risk.
Patient education about these rare but frightening side effects is essential to improve medication adherence and better outcome of the management of the mental disorder.
Patient perspective:
“The first hallucination I had was one hour roughly after taking lithium and lamotrigine. It was the Pope which appeared as bright light on my wall. He was wearing a white gown and he had gold jewellery. The picture was so clear and very detailed. I’m not religious and this image would not be something I would think of.
Every day on lamotrigine I had black spots moving quickly around the walls. They were in size of about an inch, 20-30 moving around at one time. Like spiders but without legs. I was really scared at first because I hate spiders. It was very unsettling and I changed my whole bed, away from my wall, and had trouble sleeping.
There was another night when I had similar to the black dots, where instead I had smaller black dots like bees moving into the corner of my room. They were all slightly moving as if they were getting their places. There were hundreds of them.
The scariest incident that happened was hearing voices downstairs. I was so sure that people had broken into the house; I went downstairs armed with razors. I was going to cut DNA from the burglars to give to the police as evidence. When I checked the house, there was no one there.
When I was taking lamotrigine with the lithium, it made me very unsettled, more anxious and mentally unstable. I was so tiered for not sleeping and my decisions irrational. It wasn’t a pleasant place to be for me personally.”
Hyperthyroidism is a common endocrine disorder and is mainly treated with anti-thyroid medications like propylthiouracil (PTU) and carbimazole. These medications have a large number of adverse effects, the commonest being skin rashes, and some are rare like agranulocytosis. Vasculitis is uncommon, but ANCA positivity is reported more in propylthiouracil and rarely with carbimazole or methimazole (1).We report a female patient with Graves’ disease who developed ANCA associated vasculitis while on carbimazole treatment.
Case report
A 29 year old female Filipino patient came to us with history of palpitations, tremors and weight loss for the last one month. Her thyroid profile showed severe hyperthyroidism (TSH <0.005, FT3-11.5, FT4-45.6) She was diagnosed with Graves’ disease as her anti-TSH receptor positive and was started on carbimazole 10mg tds. After three weeks of treatment, she developed macular rash over arms and legs and swelling of small joints of both hands. She noticed pain and colour change of both the hands and experienced typical Raynaud’s phenomenon. She had no renal or lung involvement.
On examination her blood pressure was 120/84mmHG, pulse 104 beats per min, temperature -37.1̊C. She had a mild diffuse goiter. Her X-ray chest, ECG and urine dipstick routine were all normal. Her CRP and ESR were raised. X-rays of the hands were normal. P-ANCA was positive. Antimyeloperoxidase antibody was positive. Anti-TPO and TSH receptor antibodies were positive.
Diagnosis of carbimazole induced vasculitis was made. The patient was treated with prednisolone 40mg daily once daily which was tapered over three weeks. She improved within 48hours and was asymptomatic after three weeks. She was treated successfully with radioiodine ablation. Her MPO-ANCA after 6 months was negative.
Figure 1. Pictures of the hands showing Raynaud’s phenomenon
Figure 2. Pictures of the hands showing Raynaud’s phenomenon
Discussion
ANCA positive vasculitis in association with antithyroid drugs was first reported in 1992 (2).There has been 32 cases of ANCA positive vasculitis associated with antithyroid medications reported up until now (3). The presenting symptoms are variable and may include renal involvement (67%), arthralgias (48%), fever (37%), skin involvement (30%), respiratory tract involvement (27%), myalgias (22%), scleritis (15%) and other manifestations (18%) (3).
In these patients the underlying thyroid disease is most commonly Graves’ disease but ANCA positive vasculitis has also been seen with association with toxic multinodular goitre (4). Recent studies have shown high frequency of ANCA positivity in patients with Graves’ disease treated with antithyroid medications, especially with PTU. Most cases of ANCA positivity are seen in patients on long term therapy (greater than 18 months) or in those with recent commencement of therapy as seen in our patient. However, a small percentage of these go on to develop features of vasculitis (3).
The majority of cases of vasculitis (88%) have been reported in association with PTU, vasculitis associated with carbimazole is very rare (5, 6, and 7). The pathogenesis of ATD associated vasculitis is not clearly understood. PTU has been shown to accumulate within neutrophils (8) and bind myeloperoxidase (9). The binding alters the configuration of myeloperoxidase (9) and may promote formation of autoantibodies in susceptible people. There has been no data with regards to whether carbimazole can alter the configuration of myeloperoxidase. ANCA positive vasculitis may be more common in patients of Asian ethnic origin, with half of cases reported from Japan (3). Our patient was from Philippines.
Wadw et al have reported 25% of patients were positive for MPO-ANCA in PTU group whereas in methimazole group 3.4% were positive (10).
This case highlights the awareness of this relatively rare adverse effect of a thyroid medication which may lead to fatal renal and pulmonary complications. Early diagnosis and withdrawal of the offending medication is important. In asymptomatic patients the significance of ANCA positivity is not clear but early definitive therapy in the form of radioiodine ablation or surgery should be considered.
A 44-yr-old patient with history of ileal Crohn’s disease was admitted to our Department because of asthenia, subclinical jaundice, painful hepatomegaly, fluid retention and ascites. In 2008 the patient was diagnosed with bladder cancer and was treated by surgical resection of the cancer and intravesical chemotherapy with mitomicyn C. In 2010 he was given azathioprine (AZA) at 2 mg/kg for Crohn’s disease and 3 months later he developed an increase in serum alkaline phosphatase, gamma-glutamyl transpeptidase and transaminases. He was then started on 1.5 mg/kg 6-mercaptopurine (6-MP) once daily. After 9 months he stopped 6-MP because of nausea, vomiting and abnormal liver function tests; 6-MP was therefore discontinued until the normalisation of markers of liver function. Two months later, when the transaminases were within the normal range, he received 6-thioguanine (6-TG) 25 mg a day, that was progressively increased to 80 mg a day. Three months later, the patient was referred to our Department with painful hepatomegaly, ascites and asthenia. Laboratory tests on admission revealed an elevation in AST 198 U/l and ALT 209 U/l. Total bilirubin was 3 mg/dl (direct bilirubin 1.5 mg/dl), LDH 784 U/l, alkaline phosphatase 191 U/l and ammonia 112 umol/l. Virological markers (HBsAg, HBcAb, anti HCV, HBV DNA) were negative. Patient was apyrexial, showed normal blood pressure (130/80 mmHg), tachycardia (110 bpm) and 97% SaO2 on room air. Physical examination revealed right hypochondrial tenderness, abdominal distension and shifting dullness, suggesting the presence of ascites. The rest of the physical examination was unremarkable. An echo-Doppler evaluation revealed thin linear suprahepatic veins and confirmed the presence of ascites. A CT scan of the abdomen showed hepatomegaly with dishomogeneous enhancement after dye injection (mosaic pattern). There was no evidence of any venous thrombosis or splenomegaly (Figure 1A); 6-TG was withdrawn empirically and the patient was started on therapy with albumin 25 g/day and spironolactone 200 mg/day. The average serum Na+ level during diuretic treatment was 134 mEq/l. An abdominal paracentesis of two litres was necessary, due to the progressive increase of ascites.
FIGURE 1A. CT scan of the abdomen on admission: Dishomogeneous enhancement of the liver after dye injection (mosaic pattern) (arrow). Suprahepatic veins are not detectable.
FIGURE 1B. Histological pattern of the liver biopsy specimen: marked centrilobular congestion (arrows) with hepatocyte dropout. There is no evidence of centrolobular veins thrombosis.
A routine laboratory investigation of ascitic fluid showed < 500 leukocytes/µL and < 250 polymorphonuclear leukocytes (PMNs)/µL. The ascitic fluid total protein level was 2.1 g/dl and serum-ascites albumin gradient (SAAG) was > 1.1 g/dL. No neoplastic cells were found. A transjugular liver biopsy was then performed, showing marked centrilobular hemorrhage with hepatocyte necrosis. There was mild ductular reaction, with no evidence of centrilobular vein thrombosis. The histologic diagnosis confirmed veno-occlusive disease (VOD) (Figure 1B). Screening for thrombophilia was also done, showing low levels of serum protein C and protein S. There was no mutation of JAK-2 V617F. The patient was then treated with a hyposodic diet, mild hydric restriction, enoxaparin,spironolactone, lactulose and omeprazole. He was discharged two weeks later, and after 3 months a complete regression of ascites and hepatomegaly occurred, and echography of the liver was unremarkable (Figure 2A and 2B).
FIGURE 2A. Echography of the liver at follow up. No evidence of ascites.
FIGURE 2B. Echography of the liver at follow up. No evidence of ascites. Suprahepatic veins are detectable (arrow)
Discussion
Although VOD was known among complications of 6-TG in childhood, this case-report emphasises the occurrence of VOD in adults with Crohn’s disease, as first described by Kane et al. in 20041. The thiopurine drugs were developed more than 50 years ago, and 6-MP was first used as a drug in 19522. Since then, 6-MP and 6-TG have been widely used to treat acute lymphoblastic leukemia in children. VOD mimicking Budd-Chiari like disease was then described as a frequent complication of 6-TG in pediatric patients given the drug for lymphoblastic leukaemia. Later on, in 1976, Griner et al. described the cases of two adult male patients with acute leukaemia developing a fatal Budd-Chiari-like disease while receiving 6-TG3. Since patients were given 6-TG plus cytosine arabinoside, authors were unable to ascribe this complication solely to 6-TG3. VOD exclusively related to 6-TG was first described by Gill et al., who observed a clinically reversible liver VOD developing in a young man with acute lymphocytic leukemia after 10 month administration of 6-TG4. Furthermore, sinusoidal obstruction was also reported in a patient with psoriasis treated with 6-TG and other cytotoxic therapy5. In 2006, a European 6-TG Working Party established that 6-TG should be considered a rescue drug in stringently defined indications in inflammatory bowel diseases (IBD). The indication for administration of 6-TG should only include its use for maintenance therapy as well as intolerance and/or resistance to aminosalicylates, azathioprine, 6-mercaptopurine, methotrexate and infliximab. Moreover, 6-TG must be withdrawn in case of overt or histologically proven hepatotoxicity6. Although Ansari et al 7 found no nodular regenerative hyperplasia (NRH) in the liver of patients given 6-TG, Dubinsky et al.8 described NHR as a common finding in 6-TG-treated patients with inflammatory bowel disease in the absence of VOD. By contrast, in our case report we showed histological pattern of VOD and, in accord with Gisbert et al.9, would suggest that 6-TG should not be administered out of a clinical trial setting. Given that the proportion of patients with Crohn’s disease achieving an improvement of symptoms during 6-TG treatment is similar to that after methotrexate10 or infliximab6, these drugs should therefore be considered as second line therapy in patients intolerant or resistant to azathioprine and 6-mercaptopurine.
A 41 year old female patient (ASA II) underwent an incision and drainage of her perianal abscess under a general anaesthetic as an urgent procedure. She was known to have anorexia nervosa and was under medical management for it. She had a BMI of 18.5. She also suffered from eczema and mild asthma. She gave a history of irregular heart rhythm in the past. She had a normal ECG and echocardiogram. She was on fluoxetine, salbutamol inhaler, beclometasone inhaler and ricatriptan. She had normal blood investigations prior to induction.
Her anaesthetic was induced with propofol and fentanyl and was maintained on oxygen/ air/ sevoflurane. She was on spontaneous ventilation through a laryngeal mask. She also received paracetamol and ondansetron intraoperatively. She was haemodynamically stable during the twenty minute procedure, which was done in the lateral position.
The laryngeal mask came out ten minutes after her arrival in recovery. The patient asked for pain relief ten minutes after waking up. IV pethidine 25mg (diluted to 12.5 mg/ml) was given by the recovery nurse who, within five minutes, noted severe redness in the distribution of the vein into which it was injected (Figure 1). The anaesthetist was notified, who then flushed the IV line with normal saline. The redness settled down within 15-20 minutes of the start of the reaction ( Figure 2 to 4). The patient was haemodynamically stable and didn't complain of any local or systemic symptoms.
Discussion
Pethidine has been known to release histamine on systemic administration1. It can also have interactions with various drug groups like SSRIs and MAO inhibitors to cause serotonin syndrome2,3 and can present with tachycardia, hypertension, hyperthermia, agitation and even seizures, among other signs and symptoms. Pethidine is equipotent to morphine and codeine in terms of histamine release 4.
This case is most likely due to profound histamine release in a patient with atopic tendency. The factors thought to increase the incidence and severity of this reaction are 5:
Old age
Thin body structure
Poor peripheral circulation
Volar > dorsal veins
Repeated injection into the same superficial vein
High concentration of solution of injection (>10 mg/ml solution)
The factors that have no influence are:
Pretreatment with an antihistamine
History of previous pethidine use
Using pethidine as a premedication
In the past, diluting pethidine with 0.25% procaine also provided protection against the reaction.
There were no other signs of serotonin excess in this patient and she came to no harm. The presentation was dramatic enough to cause concern but was self-limiting.
Metastatic carcinoma to the sinonasal tract is rare. We describe a patient with an aggressive follicular variant of papillary thyroid carcinoma who presented with an unusual metastasis to sphenoid sinus.
Case report
A 44 year old Hispanic woman presented at Queens Hospital Center in June 1988 with airway obstruction and was found to have a 10x12 cm firm mass in the left thyroid lobe, and palpable left supraclavicular node. She had no prior history of radiation, and no family of thyroid cancer. She underwent a total thyroidectomy with a modified radical neck dissection. Pathology revealed a follicular variant of papillary thyroid carcinoma: non-tall cell variant. Six of fifty (6/50) lymph nodes were positive. Post-surgery, patient received Iodine-131 ablation therapy (93 mCi) and was placed on thyroid hormone suppressive therapy. Non-stimulated thyrogen total body scan a week after therapy was negative. Thyrogen was not available at that time.
The patient was non-compliant with thyroxine and thyroid stimulating hormone (TSH) was often elevated (13-80 mlU/ml). However, the serum thyroglobulin remained less than 5.0 ng/ml and antithyroglobulin antibody was negative. A repeat total body scan (with 5 mCi I131) 6 months later and 4 years later with thyroxin withdrawal (TSH 36 mIU/ml and 48 mIU/ml respectively) was negative, and patient was continued on thyroxine suppression therapy.
Five years after initial presentation, the patient developed urinary retention and lower extremity weakness. A myelogram revealed block at T1-T2. Patient underwent laminectomy. Pathology revealed metastatic follicular variant of papillary thyroid carcinoma. Since iodine containing contrast was used during the myelogram, I131 iodine therapy was not given. External radiation of 2000 CGY to C7-T5 was administered.
A total body scan 8 weeks post laminectomy (when 24 hour urine iodine < 100 microgram/litre, and TSH was 38 mIU/ml after thyroid hormone withdrawal) was negative, the thyroglobulin level was 5 ng/ml and negative antithyroglobulin antibody (at that period of time, positron emission tomography (PET) scan was not an available option). For the next 2 years of follow up, the patient was maintained on thyroxin suppression therapy, this time with good compliance (TSH 0.1 mIU/ml, thyroglobulin less than 5 ng/ml and negative antithyroglobulin antibody). She did not show up for follow up lumbar computerised tomography (CT).
Seven years after the initial presentation, she complained of headache and double vision, and a three month history of amenorrhea. The thyroglobulin at this time was elevated (20 ng/ml). Chest X-ray was positive for two nodules in the right lung. Magnetic resonance imaging (MRI) revealed a soft tissue mass in the sphenoid sinus, eroding the basi-sphenoid and extending into the nasopharynx (Fig. 1 ABCD). The mass also eroded the sella floor displacing the pituitary gland upwards (arrows). Bone scan revealed focal abnormalities in the upper thoracic spine, ethmoid bones and base of the skull. At that period of time, PET scan was not an available option. Pituitary function testing revealed TSH 0.1 mIU/ml, free T4 level 1.2 mIU/ml. AM cortisol 5.3 mcg/dl, prolactin 182 ng/ml, ACTH 12 pg/ml, FSH 11.5 mIU/ml, LH 4.0 mIU/ml, and Estradiol 20 pg/ml.
Figure 1: A-T1 weighted midline sagittal MRI scan without contrast. B-T1 weighted midline sagittal MRI scan with contrast. C-T2 weighted axial MRI scan through the lesion. D-Axial CT scans without (on the left) and with (on the right) contrast. Note: The large destructive and enhancing lesion (*) in the sphenoid sinus associated with destruction of the basisphenoid, clivus and sellar floor. Note the normal pituitary gland (arrow) is displaced upwards out the sellaturcica.
Biopsy of the sphenoid sinus mass confirmed that it was metastatic papillary thyroid cancer, follicular variant. The tumour cell nuclear DNA was diploid and P53 and K167 were negative (Impat, NY). The patient was placed on hydrocortisone replacement and continued on thyroxine suppression therapy. Three months later the patient suffered a cardiorespiratory arrest and expired.
Discussion
Metastasis to the sphenoid sinus is rare from any tumour, and from papillary thyroid cancer it is extremely rare. An extensive world literature review revealed only 4 cases of spread to sphenoid sinus region from papillary thyroid cancer.1-4
Renal cell carcinoma is the most common tumour of paranasal sinus metastasis, 41.8%. The average age is 58 years, with slight predominance of males. The most common presentation was epistaxis, 31%. The most common causes of sphenoid metastasis are gastrointestinal and renal tumours5.
Von Eiselsberg et al. in 1893 described one case of metastasising thyroid carcinoma to sphenoid sinus.6 Harmer et al., 1899, reported a case of medullary thyroid carcinoma metastasis to sphenoid/ ethmoid sinus and nose. 7 Barrs et al. in 1979 reported a case of metastasis of follicular thyroid carcinoma to sphenoid sinus and sphenoid bone.8 Chang et al. in 1983 described a case of metastatic carcinoma of the thyroid to the sphenoid sinus.9 Renners et al. in 1992 reported one case of metastasis of follicular thyroid carcinoma to the paranasal sinuses, including the sphenoid sinus. 10Yamasoba et al. in 1994 reported a case with follicular thyroid carcinoma metastasising to sinonasal tract which also included sphenoid sinus.11 In the same year, Cumberworth et al. reported a case of metastasis of a thyroid follicular carcinoma to the sinonasal cavity which head CT showed sphenoid, ethmoid, frontal and maxillary sinuses. 12In 1997, Altman et al. described a case of follicular metastatic thyroid carcinoma to paranasal sinuses which included the sphenoid sinus. 13 The reported cases of thyroid cancer metastasis to sphenoid sinus are in table 1. Four cases were papillary thyroid carcinoma (included follicular variant of papillary thyroid carcinoma), six cases were follicular thyroid carcinoma, 1 case was medullary thyroid carcinoma and 1 case was unspecified thyroid carcinoma.
Table 1: Cases of thyroid metastases to the sphenoid sinus
Author
Age
Sex
Presenting symptoms
Histologic type
Present case
44
F
Headache, double vision and amenorrhea
Follicular variant papillary thyroid carcinoma
Mandronio (2011)
53
F
Blurring of vision of left eye
Papillary metastatic thyroid carcinoma
Nishijima (2010)
81
F
Epistaxis
Differentiated papillary thyroid carcinoma
Argibay Vasquez (2005)
53
F
Headache, paresthesia in the right eye region and left monocular diplopia
Differentiated carcinoma of thyroid, follicular variant of papillary cell
Altman (1997)
81
F
Progressive headache
Follicular thyroid carcinoma
Freeman (1996)
50
M
Facial pain, proptosis of the left globe and left horner’s syndrome
Metastatic papillary thyroid carcinoma
Yamatosoba (1994)
34
F
Hearing loss in right ear
Follicular thyroid carcinoma
Cumberworth (1994)
62
F
Right nasal blockage
Follicular carcinoma of the thyroid
Renner (1984)
61
F
Profuse right unilateral epistaxis
Follicular thyroid adenocarcinoma
Chang (1983)
50
F
Intermittent epistaxis, weight loss and pain in the right nasopharyngeal region
Follicular carcinoma with papillary foci
Barrs (1979)
54
F
Progressive loss of vision in the left eye
Follicular thyroid carcinoma
Harmer (1899)
44
F
Headache
Medullary thyroid carcinoma
von Eiselsberg (1893)
38
M
Chronic meningitis
Thyroid carcinoma
Pathologic lesions involving the sphenoid sinus include inflammatory disease, mucocele, chordoma, nasopharyngeal carcinoma, plasmacytoma, primary sphenoid sinus carcinoma, adenocystic carcinoma, pituitary adenoma, and giant cell granuloma. Benign disease often presents with a more gradual obstruction and disturbance of vision. This contrasts with the acute and progressive disturbances of vision in all cases reported with malignant lesions of the sphenoid sinus.14
Our patient presented with complaints of double vision for 6 months and headache. After imaging with MRI and given her previous history of metastatic thyroid cancer, the most likely diagnosis was metastases to the sphenoid sinus from the thyroid cancer, which was confirmed by tissue biopsy. Since this patient had evidence of bone metastasis, it is likely that the tumour first metastasised to the bone and then ruptured into the sphenoid sinus. The tumour appears to have eroded the sellar floor, extending into and displacing the pituitary gland, causing secondary hypoadrenalism.
In our patient, low thyroglobulin proved to be an unreliable marker because it was low when the patient had metastasis of the tumour in the spine. These tumours are more aggressive and today, PET scanning has proved more reliable in following them, a modality that was not available at the time for our patient. The possible explanations for negative total body scans in patients with metastatic differentiated thyroid cancer are a) technical limitations of the scan in detecting the tumour cells, and b) failure of the tumour tissue to trap iodine.
There are several unusual aspects in this patient’s presentation. Firstly, the initial presentation was unusual, since this tumour was very aggressive with rare sites of distant metastases. Perhaps the long periods of hypothyroidism when patient was noncompliant promoted the aggressive nature of this tumour. Secondly, the failure of known tumour markers, i.e. serum thyroglobulin and total body scan to identify these metastases. Thirdly, our patient’s tumour cell nuclear DNA was diploid. Investigations have shown that the DNA ploidy pattern as determined by flow cytometry is an important and independent prognostic variable.15-17 Fortunately, aggressive follicular variant papillary cancer of thyroid (non-tall cell type) is very uncommon.
Generally, total body scan negative with low stimulated thyroglobulin is an excellent prognostic sign. Our patient demonstrates that we need to remain vigilant for the unusual tumour especially when the initial presentation showed so much bulky disease. The need for additional tumour markers will help to identify aggressive well differentiated thyroid carcinoma cases.
Acknowledgement
Appreciation is extended to Ms. Deborah Goss and Mr. Timothy O’Mara, librarians, in helping with literature search and preparing the manuscript. No other financial sources or funding involved in the formation of manuscript. No potential financial conflicts of interest.
Patella fractures account for 1% of all fractures but there is little in the contemporary literature regarding outcomes. It is accepted that where fixation is required it needs to be rigid and tension band wiring using cannulated screws or k-wires is the accepted standard.1 Recognised complications associated with this form of fixation occur in up to 15% of cases2 and include; infection, loss of fixation, knee stiffness, post-traumatic osteoarthritis, malunion, nonunion and irritation from hardware.3Thereis nothing in the literature regarding the natural history following fixation.
We report an unusual complication of an osteochondral defect being generated as a direct result of a malunion of a patella fracture previously fixed by a tension band wiring technique.
Case Report
A 35-year old lady presented to our unit after a direct fall on to her left knee with an associated dislocation of her patella that spontaneously reduced on extension, at the time of injury. Three years previously she had sustained a patella fracture that had been treated with tension band wiring. From the time of the original fixation she had experienced mild persistent anterior knee pain, with a reduced range of motion and grinding. She had been discharged from further follow up.
On this presentation, examination revealed that she had a marked knee effusion with a functional extensor mechanism and a range of motion from 0-60 degrees.
The initial radiographs demonstrated that she had broken hardware and an incongruency of the patella suggesting malunion on the articular surface with a residual step (figure 1). In addition, an osteochondral fragment was identified in the patellofemoral joint. Computer tomography was undertaken and confirmed the osteochondral fragment had come from the lateral femoral condyle and a spur like prominence on the articular surface of the patella (figure 2). The mechanism was that this bony spur had been driven into the articular surface of the lateral condyle on dislocation resulting in the osteochondral fragment being generated.
Intraoperative findings confirmed this and measured the osteochondral fragment as 40x15mm (figure 3). In addition it was found that the lateral longitudinal wire had protruded into the joint causing a vertical linear defect in the articular surface of the trochlea.
The osteochondral defect was reduced and stabilized with interrupted 3/0 PDS sutures achieving a smooth articular surface (figure 4). In addition a patelloplasty and a lateral release were performed to remove the bony prominence and restore patella tracking respectively. At 6-month follow up this patient was progressing well with rehabilitation and the majority of her chronic symptoms had resolved.
Figure 1. Lateral radiograph demonstrating an incongruency of the patella suggesting malunion on the articular surface with a residual step
Figure 2. Computerised Tomography confirming an osteochondral fragment that had come from the lateral femoral condyle.
Figure 3. Osteochondral fragment from the lateral femoral condyle measuring 40x15mm.
Figure 4. The osteochondral defect stabilized with interrupted 3/0 PDS sutures achieving a smooth articular surface
Discussion
Although patellae account for 1% of fractures their functional outcome remains largely ignored in the literature. This case presents an unreported complication and highlights that symptoms can remain following the initial fixation that are accepted either by the patient or the treating centre and not further investigated.
Osteoarthritis of the knee remains the most common musculoskeletal complaint in general practice but even then only a third of those with symptoms seek medical advice. Therefore the lack of re-referrals following fixation is not an accurate way to assess outcome following patella fractures treated with this mode of fixation.
Patella fractures represent only a small number of fractures and therefore assessment of treatment and outcomes is problematic, particularly as there is no standardised rehabilitation regimen.
We report on this case as it illustrates a complication of patella fracture fixation that has not been previously described or routinely recognised and, additionally, highlights the fundamental importance of a comprehensive standardised post-operative imaging follow-up regimen. It may be that patients are not currently being correctly counselled regarding the longer-term expectations following patella fracture. A study to define the natural history of patella fractures following contemporary management is needed.
Diabetic patients with peripheral neuropathy are predisposed to foot injury. In Asian countries, a common culture among patients with peripheral neuropathy is to immerse their feet in hot water baths, with a belief that it will “improve circulation” and hence “cure the numbness”. We hereby report three cases of severe burn injuries of the feet presented to our hospital over a span of six months due to the above belief.
Case Report
The first patient was a 53-year old Malay gentleman with poorly controlled diabetes mellitus for six years, complicated with peripheral neuropathy, diabetic nephropathy and right eye cataract (latest HbA1C 8.1%), treated with oral anti-diabetic agents. He had a habit of using hot footbaths for numbness of both feet. Two weeks prior to presentation, due to increased feeling of numbness, he immersed his right foot into a self-prepared tub of hot water with added salt, followed by application of traditional sea cucumber gel. That evening, he noticed blistering of his right foot. Despite advice for admission, he chose to do the dressing as an outpatient in a local clinic. He presented two weeks later due to a worsening wound. At presentation, 4% full thickness burn of his right foot was noted, complicated by secondary infection (Figure 1). He underwent wound debridement, and subsequent split skin grafting. He had a prolonged hospitalization of five weeks due to secondary pseudomonas wound infection requiring parenteral antibiotics.
Figure 1. Right lower limb upon presentation to our hospital
The second patient was a 26-year old Indian gentleman with type I diabetes mellitus for nine years, complicated with diabetic nephropathy and peripheral neuropathy. His wife usually prepared hot water footbaths for him to improve his feet circulation. He developed 5% full thickness burn when he immersed his right foot into a pail of boiling water, not knowing that his wife had not added cold water into the footbath. He presented himself after two days and was hospitalized for two weeks. He recovered after wound debridement and split skin grafting.
The third patient was a 17-year old Chinese lady with poorly controlled type I diabetes mellitus for eight years, complicated with diabetic nephropathy (latest HbA1C 10.0%). She used hot water steam therapy with an aim to cure her recent onset of left foot drop, but was unaware of the temperature of the water. She developed blisters on her left foot, but only presented herself two weeks later when she developed left foot gas gangrene. She had a prolonged hospital stay of eight weeks with recurrent hospital acquired infections, including Methicillin-resistant Staphylcoccus aureus (MRSA). Despite multiple wound debridement, she required amputation of her left fifth toe (Figure 2).
Figure 2. Left lower limb post Ray amputation
Discussion
Peripheral neuropathy is a known complication of diabetes mellitus. More than 50% of patients who are over 60 years old have this complication.1, 2 Thermal injury to the feet in patients with neuropathy has been reported after walking barefoot on hot surfaces3 and after application of hot water bottles or heating pads during winter months.4, 5 The use of thermal footbath as a cause of burn injury is mostly due to patient-misuse or ignorance of correct usage.6, 7 In contrast, in Asian countries, a common culture among patients with peripheral neuropathy is to immerse their feet in self-prepared hot water without checking the water temperature,8 with a belief that it will “improve circulation” and hence “cure the numbness”. This practice has led to accidental burn injuries as described in our case reports.
There are a few reasons why patients with diabetic peripheral neuropathy end up with such a severe complication after the use of thermal footbath. Firstly, the temperature of the thermal bath may be underestimated. The time to develop full thickness burn reduces exponentially with just minimal increments in water temperature.9 Secondly, lack of pain despite the burn can prolong exposure to the heat source. In addition, concomitant peripheral vascular disease and endothelial function can limit vasodilatation to conduct heat away hence further aggravate the thermal insult.
Another important contributing factor of complicated wounds are the delays in seeking treatment as the result of lack of pain despite the burn injury. In a study done by Memmel et al on 1794 patients (of which 130 were diabetics) who presented with burn injuries, the majority of non-diabetic burn patients (63%) presented within 48 hours of injury, but only 40% of diabetic patients sought treatment within that time frame. Significantly more patients with diabetes presented after two weeks compared to those without diabetes. As burn injuries are highly susceptible to secondary infection, any delay in presentation further complicates and prolongs hospital stay.10,11 Not surprisingly, our two patients who presented two weeks after their burn injury had a prolonged and complicated hospital course compared to our second patient who presented soon after the burn injury. Increased susceptibility to infection and delayed wound healing from poor circulation contribute to prolonged recovery and poorer clinical outcomes in patients with diabetes mellitus, with some needing amputation as noted in our third patient.
As a healthcare provider we play a role in preventing this misfortune. Routine screening for the presence of peripheral neuropathy and vascular disease should be done during clinic visits to identify high-risk patients. Specific education regarding avoidance of thermal footbath and consequences of this highly preventable injury should be incorporated into standard diabetic foot care education. If patients choose to immerse their feet in hot water, temperature of the water should always be measured with a thermometer and immersion time should be limited. If a wound develops, they should present early to hospital for immediate treatment.
Conclusion
Thermal footbath for therapeutic purposes is commonly practiced in Asian culture. Our case reports highlight the serious consequences of this practice in diabetic patients with peripheral neuropathy. More public awareness and patient education is needed to prevent these injuries and to avoid the high cost of prolonged hospital stay and losses to the patient.
Bortezomib is a reversible proteasome inhibitor, currently approved by US FDA for use in multiple myeloma and mantle cell lymphoma. It has been shown to cause new onset and exacerbation of underlying congestive cardiac failure (CHF) in some case reports. Although the exact mechanism of bortezomib induced congestive cardiac failure is unknown, studies have shown dysregulation of ubiquitin proteasome system (UPS) in human cardiac tissues in end stage heart failure1-3. Furthermore, a study in rats has shown reduced left ventricular contractility after bortezomib administration, which was attributed to reduced ATP synthesis in mitochondria of cardiac myocytes4. Our case demonstrates new onset severe reversible left ventricular systolic dysfunction after 4 cycles of bortezomib in a 58 year old female with multiple myeloma. It highlights the importance of monitoring cardiac function in patients receiving bortezomib.
Case Report
A 58 year old female with past medical history of well controlled hypertension presented with severe low back pain, anorexia and unintentional weight loss of around 20 pounds over a period of 3 months in medical clinic. On evaluation of her routine laboratory tests, she was found to have haemoglobin of 6.5 g/dl, haematocrit of 19.9%, white blood cell (WBC) count of 3.9 x 103/cc, red blood cell (RBC) count of 2.18 x 106/cc and platelet count of 1.52 x 105/cc. Her blood urea nitrogen and creatinine was 10 mg/dl and 0.7 mg/dl respectively and corrected calcium level was 10g/dl. On liver function test, her total protein was 12.4 g/dl and albumin level was 2.8 g/dl. X-ray of lumbosacral spine revealed a compression fracture at the level of T12and L2 vertebra. Bone survey confirmed diffuse osteopenia, severe collapse of the body of T12 and partial collapse of L2 and L3. Due to the presence of severe anaemia and compression fractures, multiple myeloma was suspected. Urine protein electrophoresis showed two monoclonal protein bands with concentration of 46.8% and 4.8% and urine immunofixation showed two intact monoclonal IgA-Kappa immunoglobulin bands. Beta-2 microglobulin level was 5.49. Bone marrow aspiration and biopsy confirmed the diagnosis of multiple myeloma. Patient was staged as IIIA according to Durie-Salmon staging system.
Subsequently, patient was planned to be treated with eight cycles of bortezomib and dexamethasone, with bortezomib being given on day 1, 4, 8 and 11 of each cycle at a dose of 1.3 mg/m2 body surface area. Prior to initiation of chemotherapy, she received radiotherapy to spine as well. However, after completing the fourth cycle of bortezomib/dexamethasone, she was admitted to the hospital with generalized weakness, nausea and vomiting. Chest X ray revealed possible right lower lobe infiltrate or effusion along with increased bronchovascular markings and she was treated with antibiotics for suspected community acquired pneumonia. However, an echocardiogram was obtained due to bilateral crackles on physical exam and increased bronchovascular markings on chest X ray, which revealed dilation of left ventricle with left ventricular ejection fraction of 30-35%, diffuse hypo kinesis of left ventricle, mild mitral and tricuspid regurgitation and diastolic dysfunction with abnormal relaxation(Tajik grade I). Left ventricular septal and posterior wall thickness was 0.8 cm. Infiltrative Cardiomyopathy in the setting of multiple myeloma was unlikely due to the absence of bi-atrial enlargement, pericardial effusion and thick bright myocardium on echocardiogram. Cardiology consultation was sought and their impression was new onset left ventricular dysfunction due to bortezomib therapy.
Patient did not receive any further cycles of chemotherapy due to cardiotoxicity and was on optimal medical management for heart failure with lisinopril, carvedilol and isosorbide dinitrate. An echocardiogram was repeated four months after discontinuation of bortezomib, which revealed normal left ventricular contractility with global left ventricular ejection fraction of 55% and trace mitral regurgitation.
Currently, at 2 year follow up, her echocardiogram shows global left ventricular ejection fraction of 65%, trace mitral and tricuspid regurgitation and diastolic dysfunction with abnormal relaxation(Tajik grade I).
Discussion and Review of Literature
Botezomib is a novel proteasome inhibitor which acts by inducing bcl-2 phosphorylation and cleavage, resulting in G2-M cell cycle phase arrest and apoptosis5. US Food and Drug Administration (FDA) have approved bortezomib for use in multiple myeloma and mantle cell lymphoma. The common adverse effects of bortezomib observed in clinical trials and post marketing surveillance include thrombocytopenia, neutropenia, hypotension, asthenia, peripheral neuropathy and nausea. US package insert for bortezomib states that acute development or exacerbation of congestive heart failure and new onset of decreased left ventricular ejection fraction have been reported, including reports in patients with no risk factors for decreased left ventricular ejection fraction and it is recommended to closely monitor patients with risk factor for, or existing heart disease.
The role of ubiquitin proteasome system (UPS) in heart failure has been studied extensively in recent years. Two studies by Hein et al and Weekes et al in 2003 have shown presence of increased amount of ubiquitinated proteins and substrates in cardiac tissues of heart failure patients, indicating reduced activity of UPS in end stage heart failure1-3. Another study has shown impaired proteasome activity in hypertrophic and dilated cardiomyopathy likely secondary to post translational modification of proteasome6.However, in early stage heart failure, there is increased activity of UPS, resulting in remodelling and high cardiac output2. Bortezomib, by inhibiting UPS, would lead to accumulation of ubiquitinated proteins in cardiac myocytes, similar to that seen in end stage heart failure. A study in rats exposed to bortezomib alone showed development of left ventricular systolic dysfunction by echocardiography and reduced synthesis of ATP was observed in the mitochondria of cardiac myocytes4. However, the exact mechanism of bortezomib induced systolic dysfunction in humans is not clear.
There have been a few reported cases of bortezomib induced congestive cardiac failure in literature (Table 1). The amount of bortezomib administered before development of symptoms of heart failure was 20.8 mg/m2 in four patients, 3 mg/m2 in one patient and 10.4mg/m2 in one patient. Three of them have received prior anthracycline based chemotherapy. Complete reversibility of heart failure after discontinuation of bortezomib was documented only in two cases by follow up echocardiograms and brain natriuretic peptide levels7, 8. The patient described in our index case had well controlled hypertension and no additional cardiac risk factors at baseline. She developed non-specific symptoms, including weakness, nausea and vomiting after the fourth cycle of chemotherapy and was admitted to the hospital for community acquired pneumonia. However, an echocardiogram was obtained due to pulmonary congestion, which uncovered the diagnosis of left ventricular systolic failure. The two echocardiograms obtained at a follow up of 4 months and 2 years showed gradual improvement in ejection fraction to 55% and 65% respectively from 15% after chemotherapy with bortezomib.
We did a review of major clinical trials of bortezomib in patients with multiple myeloma, Waldenstrom’s macroglobulinemia and plasma cell leukaemia (Table 2) to investigate the incidence of congestive cardiac failure reported after administration of bortezomib. In APEX trial, the incidence of congestive cardiac failure was 2% in both bortezomib and high dose dexamethasone group 11. In a study on melphalan refractory multiple myeloma by Hjorth et al, 3 cases of congestive cardiac failure was reported in bortezomib-dexamethasone group and 2 cases were reported in thalidomide-dexamethasone group12. Another study evaluating the safety of prolonged therapy with bortezomib by Berenson et al reported 1 case of cardiomegaly and 1 case of pulmonary edema13. However, further studies are needed to specifically evaluate the incidence of congestive cardiac failure with bortezomib therapy.
In summary, our case and review highlights the importance of maintaining a high level of suspicion for development of congestive cardiac failure after therapy with bortezomib. Given the widespread use of bortezomib and new generation proteasome inhibitors in multiple myeloma, there might be increased incidence of new onset and exacerbation of underlying congestive cardiac failure in future. Currently, there is no guideline for routine evaluation and monitoring of cardiac function in all patients during the course of bortezomib therapy. Furthermore, it is unclear whether the severity of congestive cardiac failure is proportional to the cumulative dosage of bortezomib administration and also, if there is any correlation between onsets of congestive cardiac failure with the timing of bortezomib therapy. Further studies are required in future to address these issues.
Table 1: Review of cases of bortezomib induced congestive cardiac failure reported so far.
Author
Age/sex
Prior cardiac history and risk factors
Baseline cardiac function
Number of Bortezomib containing cycles
Exposure to other cardiotoxic medications
Amount of Bortezomib received before onset of cardiac symptoms
Lowest EF** after Bortezomib administration
EF on follow up visits
Voortman et al7
53/M
36 pack years of smoking and COPD
Echo not available; NT-Pro BNP 1389 ng/l
4
Gemcitabine
3 mg/m2
10-15% on Echo after 4 cycles
45% on MUGA scan at 6 months
Orciuolo et al9
73/M
NK*
NK
6
1 Anthracycline containing regimen
20.8 mg/m2
EF 25%
NK
Orcioulo et al9
61/F
NK
NK
4
2 Anthracycline containing regimens
20.8 mg/m2
EF 20%
NK
Orciuolo et al9
80/F
NK
NK
4
1 prior non anthracycline chemotherapy regimen received
20.8 mg/m2
EF 35%
NK
Hasihanefioglu et al10
47/M
None
EF 70% and normal coronary angiogram
2
1 cycle of Vincristine, Doxorubicin and Dexamethasone
10.4 mg/m2
EF 10%
EF 20% at 6 month follow up
Bockorny et al8
56/F
Hypertension, well controlled
NK
4
None
20.8 mg/m2
EF 20-25%
EF 55-60%
INDEX CASE
58/F
Hypertension, well controlled
NK
4
None
20.8 mg/m2
EF 30-35%
EF 55% at 4 month and 65% at 2 year follow up.
*NK: Not Known; **EF: Ejection Fraction
Table 2: Review of cases of congestive cardiac failure reported in clinical trials with bortezomib in multiple myeloma, Waldenström’s Macroglobulinemia and plasma cell leukaemia.
Authors (ref)
Study
Study population
Significant Cardiac events (n)
Berenson, J.R. et al. 200513
Safety of prolonged therapy with bortezomib in relapsed or refractory multiple myeloma
Frontline chemotherapy with bortezomib-containing combinations improves response rate and survival in primary plasma cell leukaemia
29 patients with untreated PPCL
None reported
Hjorth, M. et al. 201212
Thal-Dex vs. Bort-Dex in refractory myeloma
131 patients with Melphalan refractory MM
2 cases of cardiac failure in Thal-Dex group and 3 in Bort-dex group
Jagannath, S. et al 200916
Bortezomib for Relapsed or Refractory Multiple Myeloma
54 patients with relapsed or refractory MM
None reported
Jagannath, S. et al 201017
Extended follow-up of Frontline Bortezomib ± Dexamethasone for MM
49 patients with untreated MM
None reported
Kobayashi, T. et al. 201018
Bortezomib plus dexamethasone for relapsed or treatment refractory multiple myeloma
88 patients with relapse/refractory MM
None reported
Mikhael, J.R. et al. 200919
High response rate to bortezomib with or without dexamethasone in patients with relapsed or refractory multiple myeloma
638 patients with refractory or relapsed MM
None reported
Richardson, P.G. et al. 200320
A Phase 2 Study of Bortezomib in Relapsed, Refractory Myeloma
202 patients with relapsed MM
None reported
Richardson, P.G. et al. 200511
Bortezomib or High-Dose Dexamethasone for Relapsed Multiple Myeloma(APEX trial)
669 patients with relapsed MM
Congestive cardiac failure in 2% of each arm.
Rosino, L. et al. 200721
Phase II PETHEMA Trial of Alternating Bortezomib and Dexamethasone As Induction Regimen Before Autologous Stem-Cell Transplantation in Younger Patients With Multiple Myeloma
40 patients with newly diagnosed MM
None reported
Sonneveld, P. et al. 201222
Bortezomib Induction and Maintenance Treatment in Patients With Newly Diagnosed Multiple Myeloma
827 patients with newly diagnosed MM
Cardiac Disorders in 5% of patient in VAD group vs. 8% of patients in PAD group.
Yuan, Z.G. et al. 201123
Different dose combinations of bortezomib and dexamethasone in the treatment of relapsed or refractory myeloma
168 patients with relapsed MM
None reported
Suvannasankha et al 200624
Weekly bortezomib/methylprednisolone in relapsed multiple myeloma
29 patients with relapsed multiple myeloma
1 case of congestive cardiac failure
Conclusion
CHF is an infrequent but serious adverse effect of bortezomib. Cardiac function should be closely monitored in patients receiving bortezomib, as case reports have shown that these patients might present with non-specific symptoms like weakness and fatigue. Further studies are required to establish the frequency and mode of monitoring of cardiac function during and after bortezomib therapy.
We report a case of worsening psychiatric symptoms in a patient who was serendipitously diagnosed with Primary Hyperparathyroidism.
Behavioural change in form of aggression sometimes occurs as a component of psychiatric disorders such as psychosis, attention deficit hyperactivity disorder, autistic spectrum condition, conduct disorder and various mood disorders. It may also present as a psychiatric manifestation of an endocrine disorder such as Primary Hyperparathyroidism (PHPT)1,2.
PHPT is rare in children and adolescents with an incidence of 2-5 in 100000 3. It is characterized by hypercalcaemia and elevation of parathyroid hormone. Children with PHPT may present with non-specific complaints such as behavioural change and deteriorating school performance.
Patients who present with non-renal symptoms have a longer duration of symptoms prior to diagnosis of PHPT4,6. It seems probable that it takes much longer for diagnosis to be made in those with pre-existing mental disorder. When left undiagnosed and untreated, PHPT can be a serious disease with significant morbidity.
The finding of elevated serum calcium levels in young people is often fortuitous as they often present with non-specific symptoms3,4. A significant number of hyperparathyroidism cases with neuropsychiatric manifestation have been reported in patients without recorded pre-existing psychiatric diagnosis 3-6.
This case report highlights the need for clinicians to always consider endocrine disorder as a differential diagnosis when treating patients with psychiatric symptoms which are poorly responsive to standard treatment. It also demonstrates the relevance of an integrated approach in healthcare delivery including the importance of good communication between primary and tertiary care clinicians.
Case Report:
A 15yr old Caucasian male known to Child & Adolescent Mental Health Service (CAMHS) for management of his behavioural problems presented in crisis as a consequence of physical aggression, suicidal ideation and homicidal thoughts.
His first contact with CAMHS had been at the age of 10 when he was referred for management of his frequent aggressive outbursts. He had always been boisterous but had no previous history of significant emotional or behavioural difficulties. His developmental history was unremarkable and there were no features indicative of any neurodevelopmental disorder. There was no family history of mental illness.
His biological parents were involved in an acrimonious divorce at the time of his first referral to CAMHS so it was felt that this conflict may have contributed to his presentation.
He was referred a Child Psychotherapist for weekly sessions as the initial assessment suggested significant weakness in his attachment and identification which manifested in the instability and immaturity of mood and behaviour.
The family described minimal improvements in his capacity for self-control having had three years of psychotherapy. His behaviour remained challenging but manageable within the community until six months prior to him being re-referred by his General Practitioner (GP) for urgent psychiatric assessment.
Following parental divorce, his mum remarried but her new marriage was also turbulent and the couple had to separate. During this period of increased psychosocial stresses within his family, the patient’s behaviour escalated to a point that he was regarded as a significant risk to himself and others. It was thought that the separation between his mother and step-father might have triggered this deterioration.
The night before his urgent referral to CAMHS, he set a trap for his mother; he had put a rope around some curtains on the floor and was planning to throw another curtain over her. He also had a knife and hockey stick with him at the time. As his mother stepped into the room, he put the curtain over her head and attempted to hit her with the hockey stick. He was promptly restrained by his father, who had come to visit, before he could do much damage.
He presented as unpredictable and aggressive but would often deny recollection of any reported outbursts. He was very upset when incidents were talked about as he believed he had no control whatsoever over this behaviour – it was clear how upsetting his behaviour was to him.
He displayed uncontrollable rage on many occasions. It was usually directed at his mother and home furniture, and might last up to two hours. He appeared to seek immediate gratification and was clearly hypersensitive to his setting with a significant degree of paranoia and irritability.
He repeatedly stated that he had thoughts of wanting to kill his mother and himself especially when angry. He did not appear able to accept any responsibility for his actions, blaming his temper outbursts on his older sibling. We heard she was extremely frightened of him; he had on two occasions broken down her door.
When he came out of these rages he would become very tearful and profoundly apologetic. These difficulties had been noticed at school where his grades had been falling. He told teachers he felt suicidal and would sometimes go into the school toilet to cry especially when he thought about his inability to control himself.
Physical examination at this point was unremarkable. The Community Psychiatrist commenced him on Fluoxetine and referred him to an in-patient psychiatric unit for further psychiatric evaluation including a forensic assessment.
He was diagnosed with Asperger’s Syndrome and Attention Deficit Hyperactivity Disorder in the inpatient unit and was prescribed risperidone and methylphenidate. His GP was asked to arrange a baseline blood test, consisting of full blood count (FBC), liver function test (LFT), urea & electrolytes (U&E) and thyroid function test (TFT). There was no request for blood glucose level or serum calcium.
The GP asked for a serum calcium level estimate purely out of ‘habit’. The laboratory result showed a high level of calcium 3.89mmol/L (normal range 2.2-2.6). Based on this significantly elevated serum calcium level, a referral was sent to the Paediatric Endocrinologist.
At the Endocrinology Clinic, he described a twelve month history of generalised aches and pains in association with emotional lability. A history of fracture of his right wrist and left hallux occurring within 18 months prior to presentation was also obtained. The X-ray report showed presence of a radiolucent area in his right femur. An assay of his parathyroid hormone, Sestamibi scan and ultrasound scan of his neck were done.
The elevated parathyroid hormone level, increased serum calcium, history of fractures and X-ray features indicated the diagnosis of Primary Hyperparathyroidism. The endocrinologist was of the opinion that his PHPT has been present for a number of years. He was referred for parathyroidectomy.
His serum calcium level dropped to 2.47mmol/L two days post surgery. As calcium level normalised, his symptoms improved remarkably and his psychotropic medications were discontinued. Since then, he has successfully commenced college full time and has succeeded in obtaining good grades in his chosen courses.
Discussion:
Psychiatric symptoms cause significant impairment in children and adolescents. Having additional symptoms of hyperparathyroidism would exacerbate the psychiatric symptoms and increase the degree of impairment. This patient presented with neuropsychiatric symptoms and evidence of end organ damage which is similar to those in published reports3,4.
Research shows that diagnosis of primary hyperparathyroidism is often delayed in young people but we suspect that it may even be more delayed in those with a pre-existing psychiatric disorder as the symptoms may be more likely to be attributable to the psychiatric condition.
It is possible that the behavioural problems in this patient may have co-existed independently of each other, but the rapid resolution of the psychiatric symptoms suggests that they may have been exacerbated by hyperparathyroidism.
Our findings in this case are similar to those reported by Spivak and colleagues’ which showed that early diagnosis of hypercalcaemia can prevent unnecessary and potentially harmful treatment with psychotropic medications7.
Psychiatric diagnoses are usually formed from identification of collective symptoms some of which may occur in other medical conditions. Adopting a multidisciplinary team approach is most helpful in the management of complex psychiatric cases. This approach may encourage clinicians to take a holistic view in management of children.
It is important for clinicians to be familiar with common psychiatric symptoms and medical conditions that may mimic or cause them because the presence of non-specific symptoms in PHPT poses a significant emotional burden for affected children and their families. It is a potentially treatable condition which if not diagnosed early could lead to impaired psychosocial well-being and damage of vital organs. Parathyroidectomy has been shown to improve general health, quality of life and cognitive functioning in patients with PHPT8.
The outcome for this particular young person could have included further episodes of in-patient hospitalisation or involvement with the juvenile justice system as a consequence of further violent episodes. The achievement of adolescent milestones and his education could have been severely disrupted and may have resulted in labelling detrimental to his future.
In the current economic climate and because of the rarity of Primary Hyperparathyroidism, we do not advocate routine serum calcium estimation in all behavioural problems but clinicians should have lower threshold for screening for this condition especially in patients with worsening symptoms despite conventional treatment.
In conclusion, Primary Hyperparathyroidism should be considered in the differential diagnosis in young people with worsening neuropsychiatric symptoms which are unresponsive to standard psychiatric treatment.
Psoriasis is a common skin disorder characterized by erythematous papules and plaque formation with silver scaling. Guttate psoriasis is much less common and many studies cite a prevalence of less than 30% among patients who have psoriasis. It refers to the acute appearance of multiple skin eruptions mostly in a patient with no preexisting psoriasis and less commonly in a patient with psoriasis. We report here a case of guttate psoriasis associated with a flare of psoriatic arthritis.
Case report
A 53 year old man presented with a generalized body rash and multiple joint pains. His symptoms started a week prior to presentation. The skin rash initially appeared on his back and flanks but gradually progressed to involve the thighs and arms. He had ‘sausage fingers’, bilateral knee and ankle swelling associated with pain and sporadic metatarsophalyngeal joint pain as manifestations of his arthritis. His past medical history included hypertension, diabetes mellitus, hyperlipidemia and psoriasis with psoriatic arthritis. He did not report any recent changes in his medication. The patient denied any history of fever, sore throat, weight loss, visual problems, dyspnea, cough, gastrointestinal complaints or recent travel. Physical examination revealed a diffuse, non-blanching, pruritic, maculopapular and maculopustular rash over the trunk. He also had a scaly and diffuse erythematic rash over the lower abdomen which was non-blanching and pruritic. Auspitz’s sign was positive. He had multiple painful joints including both knees, right wrist, left proximal interphalangeal joint, and both his ankles. Left knee arthrocentesis was done which revealed joint fluid consistent with inflammatory joint disease without any evidence of crystals. Laboratory tests, including red and white blood cell count, haemoglobin, cyclic citrullinated peptide antibody, rapid plasma regain, hepatitis panel, antinuclear antibody, rheumatoid factor, lyme markers and serum uric acid revealed no abnormalities. ASO titer level was positive at 196. An x-ray of his left hand showed periarticular erosive changes along the distal aspect of the proximal phalanx. A skin biopsy was performed which revealed mild spongiosis and a perivascular lymphoplastic infiltrate. A diagnosis of guttate psoriasis was made. He was started on prednisone, methotrexate and folic acid and discharged from hospital. He was followed up in the rheumatology clinic 2 weeks after discharge and his rash had improved.
Discussion
Unlike psoriasis, guttate psoriasis is a much lesser known entity. It refers to the acute appearance of multiple skin eruptions, mostly in patients with no preexisting psoriasis and less commonly in chronic plaque psoriasis (guttate flare of chronic plaque psoriasis).
The characteristic skin lesions of guttate psoriasis are less than 1 cm in diameter, hence the name guttate (drop like). The lesions look like a shower of red, scaly tear drops that have fallen down on the body mainly involving the trunk, arms, thighs and face. Guttate psoriasis should be differentiated from diabetic dermopathy, also called shin spots, which typically begin as dull red, scaly papules or plaques and later develop into bilateral asymmetrical circumscribed shallow pigmented scars and/or brownish macular lesions with a fine scale. In diabetic dermopathy the lesions usually are greater than 4 cm in size.
Diagnosis is usually made on clinical examination; however skin biopsy is helpful in difficult cases. Histopathologic findings of guttate psoriasis vary with the age of the lesion. Findings in early lesions may be nonspecific and may include mild acanthosis, papillary dermal oedema and lymphocyte-predominant dermal infiltrate. Mature lesions exhibit parakeratosis alternating with hyperkeratosis, epidermal acanthosis, Munro microabscesses and dermal perivascular infiltrate containg neutrophils, lymphocytes and macrophages.
Streptococcal infections are well known to precipitate guttate psoriasis,1 however there have been no significant improvements in patients who were given penicillin or erythromycin when compared to those who were not treated.2 Other known precipitants are physical and psychological trauma.
The exact pathophysiologic mechanism is undetermined. The disease is believed to result from an immune reaction triggered by a previous streptococcal infection in a genetically susceptible host. Recent research points toward chromosome 6 as HLA-Cw*0602 allele positive patients are more prone to develop the guttate form.
Patients who present to physicians with altered mental health and behaviour may prematurely be diagnosed to have psychiatric illness. Psychiatric manifestations commonly accompany acute medical illnesses. As a rule of thumb, organic causes need to be ruled out before making a psychiatric diagnosis. A delay in arriving at a diagnosis could lead to prolonged psychiatric symptoms and unnecessary use of psychotropic medications. Prognosis and recovery will vary depending on the type of accompanying medical illness. Below, two cases are presented which varied significantly in terms of the sequelae.
Case 1
A female patient in her late 20’s was referred from primary care as she reported a sudden onset of “hearing voices”. The patient disclosed that she was bullied at work 6 months prior to this episode which resulted in change of job role and bullying has been dealt with. She described that the voices were talking about world politics and wanted her join MI5. She also believed that there were evil scientists who could read her mind through telepathy and felt that there were cameras and bugs planted in her house by a plumber. She became suspicious that her neighbours were spying on her. Her sleep was disturbed and she heard a ‘tick-tock ‘sound. This led to her checking pipes and walls to find the origin of the sounds. She believed that people were throwing things on to her bed and became very distressed - to the extent that the police were called on many occasions. She believed that people had been using telepathy on her and that aliens were invading the world. She was so distressed that she attempted to end her life by drinking bleach on one occasion and trying to cut her wrist on another.
There is no previous history of mental illness and no mental illness in the family.
Her medical history revealed that she had had mastoid surgery four years ago. Three weeks prior to the psychotic episode noted above, she had an ear infection with discharge from her ear. She presented to the general practitioner with psychotic symptoms at the same time. A CT scan of the brain was normal. A 2 week course of antibiotics in the form of Co-amoxiclav was given by the general practitioner. Her psychiatric manifestation resolved completely on follow up at two months without any psychotropics or Benzodiazepines, The patient was then kept under ‘wait and watch’ for six months in order to monitor any re-emergence of psychotic symptoms. She did not report any further episodes of ear infections during this period of follow up. Her diagnosis was Diseases of middle ear and mastoid (H65-H75).
Case 2
A 31 year old lady was referred to secondary mental health services from a Neurology team, presenting with psychotic symptoms following a viral encephalitis infection. A diagnosis of Herpes Simplex viral (HSV) encephalitis was confirmed by lumbar puncture and a CT scan by the Neurology team. She had made a gradual recovery from the encephalitis over a period of one month. She developed psychotic symptoms 2 weeks later, where she believed that doctors were trying to kill her and that she had been raped, with no evidence that this had occurred at any time. Her distress worsened which led to her informal admission to an inpatient mental health unit. A close assessment on the ward showed her to display an intermittent picture of worsening gait. A neuropsychiatry assessment confirmed that this lady had acquired brain injury following Herpes encephalitis. Single photon emission computed tomography (SPECT) showed extensive brain injury consistent with HSV. This lady presented with several acute episodes of psychosis where she complained of hearing voices and believed that people were trying to harm her. Her mood was labile and variably responsive to Olanzapine initially with a control of her psychotic symptoms as well as mood behaviour. She was diagnosed as suffering from Organic Delusional (Schizophrenia-like) Disorder (ICD 10 F06.2), with a picture of Paranoid Schizophrenia secondary to viral encephalitis. A maternal aunt was said to have suffered from Schizophrenia. Later on she had frequent relapses of psychosis and Quetiapine was initiated. As her symptoms did not respond to two different anti-psychotics, she was started on Clozapine which gave her reasonable stability. However, she still needs support while walking andhas been transferred to a suitable neuropsychiatric rehabilitation placement to maximise her independence and manage her ongoing needs.
Discussion
The two cases above display infective neurological diseases characterised by psychiatric presentations and greatly differing prognosis. The first case was an example of a chronic recurrent ear infection which was likely to have involved inner ear, mastoid or temporal lobe but has subsided without any long term sequelae. This was promptly treated with antibiotics at an early stage with complete recovery and there was no evidence of brain injury on imaging. The psychiatric manifestation was dramatically acute in this case and this could be partly attributable to stress at work. In particular there was neither previous history of mental illness nor a family history of psychiatric disorders.
In the second case, there was evidence of significant brain injury resulting in both physical and psychiatric sequelae following herpes encephalitis. Furthermore, this patient has a family history of schizophrenia which might have influenced the manifestation of the Mental Disorder.
There is significant evidence to suggest that childhood neurological viral infections increase the risk of psychotic illness 1. In both cases there was no suggestion of such illness in the childhood history. A recent study investigated whether HSV1 exposure was associated with structural brain abnormalities in individuals who, because of genetic or other factors, were deemed at high risk of developing psychosis. The results suggested that a history of HSV1 infection is associated with volumetric gray matter reductions in individuals at high risk for developing psychosis 2. In our second case, SPECT imaging confirmed grey matter loss and with the strong genetic risk would have led to the psychiatric illness.
The relationship between bacterial infections and psychotic illness in less well understood. Schizophrenic illnesses are often multifactorial in origin following a complex interplay between genetic and environmental factors such as infections. While there are various reports of Neurosyphilis, Mycoplasma Pneumoniae and Cryptococcal meningitis causing psychotic illness, specific bacteria could not be isolated in the first case presented 3-7. The improvement with antibiotics simply suggests that this could be a bacterial infection.
In summary, these cases clearly show the importance of identifying and treating medical illness presenting with psychiatric symptoms at the earliest to prevent long term complications.
Neuroleptic malignant syndrome (NMS) is a life-threatening neurologic disorder associated with the use of neuroleptic agents. NMS is typically characterized by a distinctive clinical syndrome of mental status change, muscle rigidity, fever, and autonomic instability. Atypical cases may present without muscle rigidity and/or hyperthermia. Association of infection in an atypical case can make the diagnosis challenging. We describe a case of NMS in a patient who presented with acute onset of altered mental status complicated with aspiration pneumonia.
Case Report
A 22-year-old female with a history of schizophrenia and seizure disorder presented with acute onset of altered mental status. Her home medications included haloperidol, clonazepam, olanzapine, trazodone, topiramate, benztropine, and trihexyphenadyl. The patient was found unarousable by her mother. She had multiple suicidal attempts in the past (overdosed on acetaminophen, drank cleaning detergent, and cut her wrist). She usually medicated herself without supervision. On examination, the patient was drowsy, afebrile (Temp 36.8oC/98.3oF), hypotensive (BP 85/64 mmHg), tachycardic (HR 105 bpm), and tachypnic (24/min). She was noted to be grunting with both inspiratory and expiratory stridor; therefore she was intubated for airway protection. Initial drug screen was negative for all substances of abuse. Acetaminophen and salicylate levels were undetectable. Head CT was unremarkable. Supportive care was provided as the patient was suspected to be overdosed with multiple medications. On the second day of admission the patient developed fever of 38.9oC/102oF. Chest xray and chest CT showed bilateral infiltrations (Fig.1), so empirical piperacillin/tazobactam and vancomycin were started for aspiration pneumonia. Sputum culture came back positive for Methicillin-resistant Staphylococcus aureus. However, the patient remained febrile with a temperature of 39.6 oC /103.2oF, despite appropriate antibiotic treatment. We suspected that there might be some other coexisting condition causing high fever. Serum creatine kinase (CK) was checked and found to be 8,105 U/L, up from 106 U/L on admission. We considered the diagnosis of NMS based on alteration of mental status, hyperthermia, autonomic instability, and elevated CK level, with the use of neuroleptic agents, although the patient did not have any muscle rigidity. The patient was started on dantrolene in addition to intravenous fluid and antibiotics. Shortly afterwards temperature and CK level started to trend down (Fig.2,3). Dantrolene was subsequently increased to maximum weight-based dose. Her mental status was gradually improved and returned to baseline. She became afebrile on day 10 of dantrolene treatment and serum CK went back to normal level after 2 weeks. Then bromocriptine was started orally and continued for 2 weeks.
Figure 1A: chest xray on admission
Figure 1B: chest xray on second day of admission
Figure 1C: chest CT on second day of admission
Figure 2. Temperature trend after starting treatment for NMS
Figure 3. CK trend after starting treatment for NMS
Atypical presentations of NMS can sometimes be difficult to diagnose, as in our patient who presented with altered mental status, fever, and coexisting infection, in the absence of muscle rigidity. We emphasize the importance of a high index of suspicion of NMS in patients using neuroleptic agents.
Discussion
NMS is an idiosyncratic drug reaction to antipsychotic medications and a potentially life threatening condition that occurs in an estimated 0.07% to 2.2% of patients treated with antipsychotics.1 Patients typically present with fever, rigidity, changes in mental status, and autonomic instability, often attributed to first generation antipsychotics, in particular after the start of medication or an increase in dosage.2 Atypical cases of NMS without muscle rigidity and/or hyperthermia have been reported, usually associated with atypical antipsychotic treatment. It has been hypothesized that atypical cases represent early or impending NMS; however pathogenesis remains unclear.3 Risk factors that have been established in case series and case-control studies include agitation, dehydration, acute medical illness, concomitant use of other psychotropic drugs, intramuscular injections and high doses of antipsychotic medications.4-6
Complications of NMS are often consequences of its symptoms. Pneumonia is the most common complication found in 13% of patient with NMS, likely due to altered mental status combined with difficulty swallowing that lead to aspiration.7 Renal failure is the second most common complication (8%), as a result of rhabdomyolysis and myoglobinuria. Other complications have been reported including myocardial infarction, disseminated intravascular coagulation, deep venous thrombosis, pulmonary embolism, hepatic failure, sepsis, and seizure.8 Mortality rate of NMS was around 20-30%.9-10 With early identification and treatment, mortality has significantly reduced to averages 10%.6
Withdrawal of the causative agent is the first step in the management of NMS. Supportive therapy, in particular, hydration, fever reduction, and careful monitoring, is the mainstay of management of NMS. In mild cases, supportive treatment alone may be sufficient.4 Adding specific therapies, such as dantrolene, bromocriptine, and benzodiazepine to supportive measures has been shown to reduce time to complete recovery, from a mean of 15 days with supportive care alone, to 9 days with dantrolene, and 10 days with bromocriptine.1 In severe cases, empirical trial of specific pharmacological agents should be started promptly. Electroconvulsive therapy is found to be effective when pharmacotherapy has failed.11
Sometimes NMS is difficult to identify in the presence of critical illnesses which can obscure the manifestation of NMS. As in our case, the patient presented with altered mental status, fever, and autonomic instability which could be simply explained by the presence of pneumonia and sepsis. However, due to lack of clinical response after appropriate antibiotic treatment, other coexisting condition was suspected. It is important to have a high index of suspicion for NMS in the setting of antipsychotic therapy. An absence of muscle rigidity should not exclude a diagnosis of NMS when the rest of the clinical picture points to this diagnosis. Elevated CK level helps support the diagnosis of NMS in patients with atypical presentation. Discontinuation of offending agent and supportive care should initiate promptly, and specific pharmacotherapy should be considered in severe cases. An early diagnosis is the key to successful treatment and patient outcome.
Conclusion
NMS is a rare but potentially life-threatening condition. Atypical presentation makes it more difficult to identify in patients with critical illnesses. Aspirated pneumonia is one of the common complications of NMS and sometimes can obscure signs and symptoms of NMS and delay diagnosis. High index of suspicion for NMS in patients taking antipsychotics is crucial. If not recognized or left untreated, NMD may be fatal.
A 34 week preterm, small for gestational age, third born male neonate to a non consanguinous married couple with father having short extremities was admitted in our NICU prematurely with respiratory distress.
On examination the baby was tachypneic with grunt and lower chest indrawing. The baby was also found to have a narrow thorax, short fingers with postaxial polydactyly in both upper limbs and right lower limb, with syndactyly in right upper and lower limb (figures 1,2,3). The cardiovascular, respiratory, abdominal and neurological examination were unremarkable with no facial dysmorphism. The fundus examination was inconclusive.
The antenatal scan showed all long bones short in configuration. The liver function tests were normal except for mild elevation of alkaline phosphatase. The Ultrasound abdomen showed hepatomegaly and no evidence of any other mass lesions. The urine examination was negative for proteinuria and haematuria. The chest x ray showed short ribs, high position of clavicle and features of hyaline membrane disease (Figure 4).
The baby was put on continuous positive airway pressure and given surfactant through an endotracheal tube twice for two consecutive days, but as the condition deteriorated, with hypercarbia and hypoxia as evident on arterial blood gases, the baby was electively ventilated with minimal settings. The baby improved and hence was extubated. After a few hours of being extubation the baby gradually developed respiratory distress and started to deteriorate. Hence the baby was reintubated. The condition of baby was explained to attenders and as the attenders were not willing to continue the treatment, the baby was discharged from hospital against medical advice and later we were informed that the baby expired within few hours after discharge from the hospital.
Fig 1 : showing long narrow thorax and short upper extremities
Fig 2 : showing postaxial polydactyly with syndactyly in upper extremity
Fig 3 : showing polydactyly with syndactyly in lower extremity
Fig 4 : chest xray showing long narrow thorax and short and horizontally oriented ribs with irregular costochondral junctions and bulbous and irregular anterior ends
Discussion
Jeune syndrome or asphyxiating thoracic dystrophy is a rare autosomal recessive skeletal dysplasia characterised by a small chest and short ribs which restrict the growth and expansion of the lungs1. The inheritance is autosomal recessive and a locus has been identified on chromosome 15q13 2. Other symptoms may include shortened bones in the arms and legs, unusually shaped pelvic bones, and extra fingers and/or toes (polydactyly)3 . It is estimated to occur in 1 per 1,00,000 -1,30,000 (again is this 130,000?) live births4.. The diagnosis is based on clinical and radiological findings. Our patient fulfills the diagnostic criteria for Jeune syndrome. The most consistent and characteristic findings were the abnormalities of the thoraxand limbs. Jeune syndrome was first described in 1955 by Jeune in two siblings with severely narrow thorax5. It is known to be genetically heterogeneous.
Several complications of asphyxiating thoracic dystrophy have been described in the literature. The respiratory problems are the main concern. A large percentage of the children with asphyxiating thoracic dystrophy die as a result of these problems. Percentages up to 80% have been mentioned in literature6,7. In our case the baby experienced respiratory distress on day one of life needing ventilator support. The thoracic malformation tends to become less pronounced with age8. A possible explanation could be the improved mechanical properties of the chest wall with growth.
Clinically, Jeune syndrome is characterized by a small, narrow chest and variable limb shortness. Associated congenital abnormalities can be postaxial polydactyly of both hands and/or feet (20%). Typical radiographic findings include a narrow, bell-shaped thorax with short, horizontally oriented ribs and irregular costochondral junctions, elevated clavicles, short iliac bones with a typical trident appearance of the acetabula, relatively short and wide long bones of the extremities, and hypoplastic phalanges of both hands and feet with cone-shaped epiphyses9. The reported case has long narrow chest, short and horizontally oriented ribs with irregular costochondral junctions and bulbous and irregular anterior ends with post axial polydactyly in both upper extremities and right lower limb with left lower limb being normal.
Jeune syndrome is sometimes compatible with life, although respiratory failure and infections are often fatal during infancy. The severity of thoracic constriction widely varies. For those patients who survive infancy, the thorax tends to revert to normal with improving respiratory function. This suggests that the lungs have a normal growth potential and the respiratory problems are secondary to restricted rib cage deformity 10.
A 54-year-old male presented to the psychosexual clinic with symptoms suggestive of persistent genital arousal disorder of 2years duration. Physical examination and investigations ruled out any underlying urological or neurological causes. He was treated with Diazepam and Pregabalin and his symptoms reduced in intensity.
Introduction:
Persistent genital arousal disorder (PGAD), also known as persistent sexual arousal syndrome (PSAS) or restless genital syndrome (ReGS), is recently recognised as a sexual health problem in western countries although it is not been considered as a physical or psychiatric disorder by DSM IV or ICD 10. PGAD is associated with constant, spontaneous and intrusive feelings of genital arousal in the absence of conscious sexual thoughts or stimuli.
The working definition of PGAD1,2 is as follows:
1) Persistent physical arousal in the genital area
2) in the absence of conscious thoughts of sexual desire or interests
3) associated with spontaneous orgasm or feelings that orgasm is imminent and
4) the symptoms not diminished by orgasm.
It may be present throughout the person’s life (primary PGAD) or develop at any age (secondary PGAD). It is associated with varying degrees of distress in the patients. This new disorder has been reported in women by numerous clinicians in the last decade. However, so far, there is only one report of two males suffering with ReGS in the literature.3 We report a case of PGAD in a male and aim to analyse the cause.
Case Report
A 54-year-old male was referred to the psychosexual clinic by an urologist with 2 years history of constant feelings of physical arousal in the genital area as if he was about to ejaculate. These feelings were associated with pain which was relieved to an extent after ejaculation. These symptoms started suddenly for the first time when he was browsing the internet and accidentally ended up in pornographic websites. But later on, the symptoms were constant without any sexual stimuli and he got some relief from attaining climax.
He described that the physical arousal in the genital area increased in intensity to a point he had to ejaculate to have some relief. He felt this “as if wanting to have climax all the time”. Post- ejaculation, he felt anxious, tired and nauseated for sometime, during which the symptoms intensified again that he needed climax. Initially this cycle repeated every 2-3 days but later on the frequency increased to 2-3 times a day. He achieved climax both by masturbation and sexual intercourse. He felt these ejaculations were unpleasant and not enjoyable. He felt frenzied if he couldn’t ejaculate and the post orgasmic feelings were severe if he avoided orgasm for a day or two. He described regular ejaculations led to less severe “come downs” but left him constantly drained.
His medical history included vasectomy four years ago with minor complication of painful scrotum which subsided fully with pain killers. He also had few urinary tract infections (UTI) in the past which were treated with antibiotics. He was initially seen by urologist who carried out physical examination which was noted to be normal. Then investigations including CT- KUB, CT- Abdomen, Urogram, Transrectal Ultrasound of prostate and seminal vesicles, Flexible Cystoscopy were done and no abnormalities noted. He also had MRI- Brain which was normal. He had no symptoms of hyperactive bladder and no varicocele was noted.
When he was seen in the psychosexual clinic, he was noted to be very anxious and expressed guilty feelings around the incident of watching pornography which initiated the onset of symptoms. There were no depressive or psychotic symptoms. Prior to attending this clinic he was prescribed duloxetine 30mgs by the urologist, which he took only for few weeks. He stopped it as there was no symptom relief. He was started on diazepam and pregabalin. The dose was increased to 2mgs qds of diazepam and 50mgs qds of pregabalin. His symptoms diminished gradually and now he remains mildly symptomatic although feeling “more in control”. He was also referred to psychologist and had an assessment. As he was not psychologically minded and unable to engage in sessions, he stopped attending.
Discussion
The clinical features in this man were consistent with the definition of PGAD. He had physical arousal symptoms, which were not related to sexual desire or thoughts and was causing severe distress to him. The symptoms were relieved by ejaculation to a certain extent. He was treated with diazepam and pregabalin which reduced the intensity of the symptoms.
There is an emerging literature on the pathophysiology, possible aetiological factors and the management options of PGAD. There are various associations reported including psychological4,5 and organic6-9 pathologies with some convincing evidence.
In this case, he suffered few UTI and a minor complication of painful scrotum following vasectomy, few years before the onset of PGAD. However he had a full urological and neurological work-up recently which didn’t show any underlying organic cause for his current symptoms. He suffered no previous depressive or anxiety disorder. Hence his current symptoms may be induced by anxiety which is further worsened by the fact that he became focussed on the genital arousal and attaining climax to relieve the pain. When he was prescribed diazepam and pregabalin, his anxiety eased and his physical symptoms diminished in intensity. However the possibility of an organic cause cannot be ruled out completely as he previously suffered sensory neuropathic pain following vasectomy. Further pregabalin is useful for both generalised anxiety and neuropathic pain. Therefore we conclude that his symptoms may be a result of interaction between physical and psychological factors. This suggests that PGAD could be a psychosomatic condition, which was already proposed as a cause for PGAD in women by Goldmeier and Leiblum.4
Similar to the causes for PGAD, there is few treatment modalities reported in the literature. These include treatment of the underlying organic causes if any found, electro-convulsive therapy (ECT) if co-morbid with mood symptoms,10 transcutaneous electrical nerve stimulation (TENS),3 cognitive behavioural therapy11 and medications like varenicline.2 We used anti-anxiety medications (diazepam and pregabalin) and achieved adequate symptom relief. This also supports the idea that PGAD could be a psychosomatic condition related to the peripheral nerves of the genito-urinary system.
This case is reported to confirm that PGAD also occurs in males, which is quite different from priapism and it could be a psychosomatic condition. More research is needed into the pathophysiology of PGAD and its management.
Trigeminal Neuralgia (TN) is relatively rare in Multiple Sclerosis (MS) affecting approximately 2% of patients1. The severity of the pain is indistinguishable whether TN is an isolated impairment or is associated with MS. However, when associated with MS, TN is often bilateral, affecting younger patients and is more refractory to medical treatment 2.
Several pharmacological agents are reported to be effective in TN associated with MS. Topiramate3,4, gabapentin5 and lamotrigine6 were all reported to benefit patients with TN associated with MS in small uncontrolled trials. Several other drugs such as phenytoin, misoprostol and amitriptyline are routinely tried in patients with TN despite the lack of convincing evidence of their efficacy7.
In 2008, Both the American Academy of Neurology and the European Federation of Neurological Societies launched joint Task Force general guidelines for the management of TN. After systematic review of the literature the Task Force came to a series of evidence-based recommendations 8. Carbamazepine and oxcarbazepine had the strongest evidence of efficacy and were recommended as the first line treatment. An earlier Cochrane systematic review reached the same conclusion. 9.
Case report
A 62 year old female patient had been suffering from MS for about 20 years. The MS presented with trigeminal neuralgia from the outset and this was then followed by pyramidal lower limbs’ weakness and sphincteric dysfunction. The patient started to use a wheelchair 10 years ago but she became totally wheelchair dependent about 6 years later.
Trigeminal neuralgia remained active throughout the 20 years. Carbamazepine (300 mg daily) provided the patient with a satisfactory control of TN. Despite having occasional break through TN pain; the patient declined having higher doses of carbamazepine as excessive sedation was an unacceptable side effect.
Recently; the patient was admitted to hospital in two separate occasions complaining of increasing malaise and confusion. Plasma sodium levels were found to be low in both occasions (first presentation 118 mmol/l and second admission 114 mmol/l). Clinical evaluation confirmed Syndrome of Anti Diuretic Hormone Secretion (SIADH) as the cause of the hyponatremia and in the absence of any other explanation for the SIADH; carbamazepine was thought to be the main reason and was duly discontinued.
Unfortunately, TN attacks came back with vengeance. During the following 6 months, therapeutic trials using gabapentine, topiramate and amitriptyline failed to show any beneficial effect on either the severity or the frequency of the TN attacks. All three drugs were duly discontinued.
The patient was started on eslicrbazepine 400 mg on a single daily dose. This dose lead to almost complete eradication of the TN attacks. The control of TN and the plasma sodium levels remained stable a year following the initiation of the therapy.
Comments
Hyponatremia, defined as a sodium level < 135 mmol/l is a common side effect of carbamazepine and oxcarbazepine therapy. The incidence of hyponatremia secondary to carbamazepine therapy ranges between 4.8 and 40 % depending on the population studied10,11. In most cases, hyponatremia is asymptomatic and continuation of the carbamazepine use is possible whilst a close eye is kept on the plasma sodium level10. In rare occasions hyponatremia is symptomatic and discontinuation of carbamazepine is warranted. Administration of demeclocycline to normalise the sodium level was suggested by some authors12 However, the long term use of demeclocycline is associated with several complications and this approach is hardly a standard practice.
Clinicians often face a dilemma when carbamazepine is the only agent able to control a specific clinical problem. With many antiepileptics available, it is unusual to face such a problem in epileptic patients. Trigeminal neuralgia on the other hand can be extremely difficult to control and carbamazepine was found to have a unique ability to manage such unpleasant condition even before its antiepileptic effects were noticed on 196213.
Eslicarbazepine is promoted as an alternative to carbamazepine when side effects occurs on otherwise responsive patients to its favourable antiepileptic effects14. Hyponatremia is rare in eslacarbazepine users with only an incidence of less than 1% in the small populations studied15,16. Frequency of hyponatraemia increased with increasing eslicarbazepine dose. In patients with pre-existing renal disease leading to hyponatraemia, or in patients concomitantly treated with medicinal products which may themselves lead to hyponatraemia17.
Our patient showed the same favourable response to eslicarbazepine as she experienced with carbamazepine. However, hyponatremia did not occur with eslicarbazepine therapy. This enabled our patient to continue with pharmacological management and avoid surgical interventions.
With the exception of epilepsy, no reports are available commenting on the use of eslicarbazepine on the wide range of conditions that carbamazepine is traditionally used for such as mental health problems and neuropathic pain. When patients are well controlled on carbamazepine whatever the indication is, the occurrence of side effects such as hyponatremia is often managed by an automatic replacement with another agent. We feel that in such patients a therapeutic trial of eslicarbazepine might be appropriate especially if the control on carbamazepine was robust or if the benefits of carbamazepine therapy were clearly superior to other pharmacological agents potentially useful for the targeted clinical condition.
Hydatid disease is due to Echinococcus species commonly granulosus sometimes multilocularis. The common locations of hydatid cyst are the liver (65% to 75%) and lungs (25%-30%) 1,2,3. Hydatid disease rarely develops in some locations such as the spleen, kidney, bones, heart, brain, peritoneum, myocardium and muscles (1-4%)1. In our review of literature, concomitant paraspinal and intrahepatic hydatid is reported rarely.
Case Report
A 25 year old male presented with backache on the right for 1 month and fullness in the right paraspinal region. No history of trauma, fever, burning micturition, pain in abdomen, weight loss or hematuria.
On clinical examination he had non ballotable lump in the right paraspinal region, measuring 15x5x5cm with ill defined margins intra muscular plane extending from the posterior subcostal margin to the iliac region with no overlying skin changes and no organomegaly.
Blood investigations including liver function tests and kidney function tests were normal. Ultrasound examination revealed hepatomegaly with thick walled cystic lesion in the right lobe of the liver and the in muscle plane in the right renal angle region.
CT abdomen (P+C) revealed a well defined hypodence non-enhancing cystic lesion measuring 45x40 cm seen in the right lobe of the liver with peripheral calcification with another lesion of similar morphology of size 14x5x3.6cm in the right paraspinal muscles with no intercommunication between them and no bone, spinal canal alteration or compression of the right kidney.
Figure 1: Right paraspinal hydatid
Figure 2: Intrahepatic hydatid
Figure 3: Coronal view of CT image
The diagnosis of paraspinal hydatid cyst was confirmed. As the patient was symptomatic for paraspinal hydatid cyst only and the size of the hepatic cyst was small, exploration of the right paraspinal region was done after 21 days of antiscolicidal treatment. There was a cyst measuring 15x5x5 cm beneath the oblique and lattisimus dorsi ,superficial to the psoas muscle without invasion into it .The cyst opened multiple daughter cysts along with pus evacuated. The cyst excised in totto without rupture & spillage. Negative suction drain was kept. Post operatively on day 3 the drain was removed, stitches removed on day 10 and discharged. Histopathology confirmed diagnosis.
Figure 4: Intraoperrative photograph
Figure 5: Photograph of multiple daughter cysts
Discussion
Hydatid disease is most prevalent in sheep and cattle-breeding areas, which is where the first step in the chain of transmission occurs. The causative agent is introduced to the dog (the primary host) through the faeces of livestock. The minute larval form of E. granulosus lives in the small intestine of the dog species. The eggs are passed in the faecesof an infected dog and can be transferred to mammal (man – intermediate host) that ingests them. After ingestion, the embryos are released from the eggs, traverse the intestinal mucosa and disseminate systemically via venous and lymphatic channels and develop into hydatid cysts in various body parts.
The cysts cannot easily grow in muscles due to their contractility and lactic acid content. The wall of a cyst in the muscle is formed by three layers: the inner germina, intermediate and outer granulomatous adventitial layer. The most common skeletal sites include hip, thigh, shoulder and humerus regions. Hydatid cysts are frequently asymptomatic1. The latent period of cyst development varies between 5 and 20 years4,5.
Serological tests are widely used to diagnose hydatid cysts. However, positive serological results do not confirm, nor negative results exclude the disease1,6. The imaging features of hydatid cysts are well described in the literature. Ultrasound scans are a sensitive, safe, non-invasive method. It is the procedure of choice for the diagnosis of cysts with a “honeycomb” pattern (type 3), as observed in our patient. Gharbi’s classification provides morphological description on ultrasound. Type 1-pure fluid collection, type 2-fluid collection with split wall (floating membrane), type 3-fluid collection with septa, type 4-heterogenous echographic pattern, type 5-reflecting thick walls.
As seen in our case the cyst fluid appears anechoic at USS, yields an attenuation value of 3-30HU at CT. Calcifications in the cyst wall as in our case are best detected on CT scans. CT has the advantage of detecting smaller cysts when located outside the liver and sometimes differentiating parasitic from non-parasitic cysts, for follow-up studies during chemotherapy. Other diagnostic means such as fine needle aspiration should be avoided because of dangerous anaphylactic reactions7.
Surgery is the optimal treatment for hydatid cyst. Open cyst evacuation is indicated for gharbi types 4 & 5, posterior cysts, central cysts, more than 3 cysts, large cysts, heavy calcification, infected cysts with above criteria, biliary communication, pulmonary communication and peritoneal rupture. Laparoscopic evacuation is indicated in Gharbi type 1 or 2, anterior cysts, peripheral cysts,1-3 cysts, small cysts, no or minimal calcification. Pericystectomy is complete resection of cyst wall without entering the cyst cavity.
Alternative therapy with non-toxic scolocidal agents or combination chemotherapy using imidazole derivatives, particularly albendazol, has been advocated in the management of patients with recurrence and high risk of contamination8.
Case Presentation: Reflex Anoxic Seizures and Anaesthesia
Reflex anoxic seizures (‘RAS’) may present, as potentially life threatening events, but these are often preventable. They are most common in preschool children (but can occur in any age) and more so in females. As a cause of seizures they are not rare; one study estimated a frequency of 8 in 1000 preschool children1, but they are often misdiagnosed. The pathophysiology of RAS is vagally mediated – a noxious stimulus causes a supranormal vagal discharge resulting in bradycardia and then astystole2. This then results in cerebral under perfusion and hypoxia. During this time the patient is often noted to become very pale with dusky lips, initially flaccid and then tonic with rigid extension and clenched jaws. They may then have a generalised convulsion, often with rolling eyes and urinary incontinence. The patient spontaneously recovers (the whole episode lasting around 30 to 60 seconds) and will feel somnolent, often remaining pale for a while.
From this description it can be easily understood how such an event can be misdiagnosed as epilepsy; however it is not associated with the uncontrolled neuronal discharge of epilepsy and if monitored by EEG this is absent2. It may also be mistaken as breath-holding attacks (where intra-thoracic pressure restricts cerebral perfusion) or Stokes- Adams attacks (where there is abnormal electrical function of the heart).
The noxious stimuli responsible can be many different things. Ocular pressure2, venepuncture3, anaesthetics4, accidental trauma and fear have all been implicated. If these stimuli cannot be prevented, management is normally just supportive (positioning, protection from trauma, oxygen) and allowing the fit to self-resolve[U1] . Further management can involve atropine5 (either acutely of preventatively), maintenance anticonvulsants6 (though these often just stop the fitting but not the syncope) and even pacemaker [U2] insertion7.
The case we encountered was that of a 20 year old female student, presenting for a planned day case removal of a molar tooth. She was otherwise fit and well with no other past medical history, only taking the combined oral contraceptive pill. Her history with RAS started at age 1, when she was admitted to hospital following two seizures. The seizures occurred every few months and she was provisionally diagnosed as suffering from epilepsy, with prophylactic treatment started. However, as she grew older she was able to describe how the attacks were not associated with a preceding aura, but rather an unpleasant stimulus (such as accidental injury). A new diagnosis of RAS was made and the antiepileptics were stopped without the seizures becoming more frequent. As she entered late childhood and adolescence the frequency of the seizures became less, but (atypically) they did not stop entirely. On preassessment she reported being seizure free for just over a year and was anxious that today could precipitate another.
After consideration, we decided to proceed with anaesthesia with the following measures. The patient was kept calm by having a clear explanation of what to expect before coming to theatre, and then was reassured by an affable theatre team (who had been informed of her condition). Atropine was drawn up and available if vagal over stimulation occurred, as was suxamethonium in case of emergency airway intervention. For cannulation, cold spray was used along with distraction. Induction was with propofol (under full monitoring) and anaesthesia was maintained with sevoflurane/nitrous oxide via LMA. To prevent pain as a potential trigger, fentanyl (at induction) and paracetamol (after induction) were given and local anaesthetic (lidocaine) was administered before any surgery. Emergence was kept as smooth as possible by removing the LMA prior to any gagging and coughing and manually supporting the airway until she was awake.
With these measures the procedure was uneventful and the patient could be discharged home as planned. We hope this case report will help improve awareness and understanding of RAS, and the steps that can be taken peri-operatively to help ensure safe anaesthesia.
In traumatic brain injury (TBI) the primary insult to the brain and the secondary insults as a result of systemic complications may result in a multitude of sequelae ranging from subtle neurological deficits to significant morbidity and mortality. As the brain recovers by repair and adaptation, changes become apparent and may result in physical, cognitive and psychosocial dysfunction. Rehabilitation is usually structured to recover physical ability, cognitive and social retraining with the aim of gaining independence in activities of daily living.
Case Report:
A 76 year old male patient was admitted to an intermediate neurorehabilitation unit following a traumatic brain injury(TBI) .He had fallen from a height of 11 feet resulting in intracerebral haemorrhage in the left parietal lobe and a left parietotemopral subarachnoid hemorrhage which was managed conservatively in the neurosurgical unit. He developed recurrent post traumatic seizures in the form of myoclonic jerks for which he was started on antiepileptic drugs (AEDs) sodium valproate ,clobazam and levetiracetam .During his stay in the acute neurorehabilitation unit, he was noted to be confused and wandering with a disrupted sleep wake cycle. Cognitive assessment showed global impairment across all cognitive domains suggesting that cognitive impairment was secondary to TBI with the chaotic sleeping pattern and fatigue having a significant effect on his cognition. He was then transferred to an intermediate neurorehabilitation unit four months post head injury for rehabilitation prior to discharge.
On admission he was confused, and disorientated. His neurological examination was normal except for mild expressive dysphasia. On the first night of his stay in the unit, he did not sleep at all, was restless, agitated and aggressive towards the staff. His initial agitation was attributed to the change of surroundings and general disorientation. However during his first week at the rehabilitation unit it was noted that his sleep wake cycle was completely disrupted .He would have short fragmented naps through the day and would regularly get agitated at night with threatening behaviour towards staff. On admission the Rancho Los Amigos scale† was 4(confused-agitated) and he needed specialized supervision. Despite environmental modification and optimal pharmacotherapy to improve sleep and decrease agitation, the patient still continued to have aggressive outbursts and no identifiable sleep wake pattern. It was noted by the nursing staff that occasionally when very agitated, the patient refused to have his night time medications including all AEDs .On such occasions he was reported to have slept better at night and did not have any daytime naps. All blood investigations were within normal limits except for mild hyponatremia with a normal creatinine clearance and CT head showed changes consistent with previous TBI with no new pathology .A neurology opinion was sought and with a Naranjo adverse drug reaction probability score†† of 7/10, a decision was taken to slowly decrease levetiracetam and wean it to stop, while continuing all other regular AEDs. The levetiracetam was reduced from an original dose of 750mg twice daily by 500mg every week with an aim to stop. This resulted in a considerable improvement in the patient’s agitation with a complete halt in the nighttime aggressiveness. His sleep wake cycle normalized and he started sleeping longer at night. His Ranchos Los Amigos scale improved from 4(confused-agitated) to 6(confused-appropriate). The patient could now participate more with the team of trained therapists in memory and attention exercises as well as regaining independence in activities of daily living.
Discussion:
TBI particularly in elderly aged over 64 years has a worse functional outcome as compared to non elderly.1Closed head injury in older adults produces considerable cognitive deficits in the early stages of recovery2 and there have been studies suggesting TBI to be a risk factor for developing Alzheimer’s disease.3Memory deficits, attention problems, loss of executive function and confusion are common after TBI.4This impaired cognitive function reduces the patient’s ability to recognize environmental stimuli often resulting in agitation and aggression towards perceived threats.TBI by itself may result in a variety of sleep disorders ranging from hypersomnia, narcolepsy, alteration of sleep wake cycle, insomnia to movement disorders. 5Sleep wake schedule disorders following TBI are relatively rare and may clinically present as insomnia.6Often these sleep disorders result in additional neurocognitive deficits and functional impairment, which might often be attributed to the original brain injury itself and thus be left without specific treatment.
While dealing with disrupted sleep pattern and agitation in the elderly following TBI, treatable causes such as neurological, infectious, metabolic, and medications should be ruled out. This is imperative as they disrupt rehabilitation and achievement of functional goals. Long duration of agitation post TBI has been associated with longer duration of rehabilitation stay and persisting limitations in functional independence.7After ruling out all the treatable causes the first focus is on environmental management with provision of a safe, quiet, familiar, structured environment while reducing stimulation and providing emotional support. The next step is introduction of pharmacotherapy to reduce agitation. Though a variety of pharmacological agents are available, there is no firm evidence of efficacy of any one class and often the choice of drug is decided by monitoring its effectiveness in practice and watching for side-effects.8In pharmacotherapy, the general principle followed is start low and go slow while developing clear goals to help decide when to wean and stop medications. Atypical antipsychotics are often used for the agitation while benzodiazepines and non benzodiazepine hypnotics such as zopiclone are recommended for treatment of insomnia.9 However atypical antipsychotics carry a FDA black box warning being associated with increased risk of stroke and death among elderly.
But what does one do when all optimal non pharmacologic and pharmacologic measures fail? That brings us back to the drawing board which in this case led the team to rethink Levetiracetam, a novel new antiepileptic that has been used as monotherapy for partial seizures and adjunctive therapy for generalized tonic clonic and myoclonic seizures. Levetiracetam treated patients have been reported to have psychiatric adverse effects10 including agitation, hostility, anxiety, apathy, emotional lability, depersonalization, and depressionwith few case reports of frank psychosis 11.While in healthy volunteers levetiracetam is noted to consolidate sleep12,in patients with complex partial seizures, levetiracetam has been noted to cause drowsiness decreasing day time motor activity and increasing naps without any major effects on total sleep time and sleep efficiency during night.13There has been an isolated report of psychic disturbances following administration of levetiracetam and valproate in a patient with epilepsy which resolved following withdrawal of valproate. 14However in practice it is used for recurrent post TBI seizures as it is a potent AED with a relatively mild adverse effect profile and no clinically significant interactions with commonly prescribed AEDs.15
Any adverse drug reaction (ADR) should be evaluated while keeping the patients clinical state in mind. This was, indeed, difficult in our case. With a history of TBI and cognitive decline, it became difficult to ascertain whether the neurocognitive issues were purely due to the nature of TBI or due to an ADR. Assigning causality to a single agent is difficult and fraught with errors. Using the Naranjo algorithm 16, with a score of 7/10(probable ADR) and a notable response on withdrawal of the offending drug as in this case helps establish possible causality.
This is a rare instance where sleep wake cycle disorder and agitation resolved following withdrawal of Levetiracetam in an elderly patient with TBI. This in turn led to the patient having a stable mood so that therapists could communicate and interact with him in order to improve basic cognitive functions such as attention, memory, thinking and executive control. This case illustrates the constant need to systematically and frequently reassess patients as they recover from TBI.
†Appendix: Ranchos Los Amigos Levels of cognitive functioning.
Ganglioneuroma is a rare, benign, neuroblastic tumour that originates from the neural crest cells. Ganglioneuroma, ganglioneuroblastoma and neuroblastoma are three maturational manifestations of a common neoplasm in the progressive order of loss of differentiation. Ganglioneuromas may be found anywhere along the line of the embryonic neural crest, from clivus to sacrum and are very rare in the pelvis. Less than twenty cases have been described in the literature with various presentations based upon location including extradural, retroperitoneal, spinal, thoracic and one solely intradural medullary location. Ganglioneuromas may stay asymptomatic for a long period and give rise to no pressure symptoms either due to slow growth leading to progressive increase in size accompanied by adaptive changes. Ganglioneuromas demonstrate long-term disease-free survival even with an incomplete surgical removal. Here we present a case of a girl aged 11 years with pelvic ganglioneuroma.
Case Report:
A girl aged eleven years was brought from a remote hilly area in Pakistan by her mother to the city hospital many miles away. She had noticed that her daughter’s lower abdomen had progressively enlarged over last few months. Her menstrual cycle was normal so the mother was concerned that despite not being pregnant, her daughter had a distended abdomen as if she was pregnant She had a good appetite and unaltered bowel and bladder function. She had no heartburn, regurgitation, nausea, vomiting, heamatemesis or melaena. She denied any bleeding par rectum, shortness of breath, cough, loss of consciousness or convulsions. Her past medical history was mundane. She had not had any surgery in the past and was not taking any medication. Examination revealed a smooth, large, fixed hard mass in the right lower abdomen and pelvis. It was palpable in the pelvis on rectal examination which was otherwise normal. Liver or spleen was not palpable and she had no ascites. Her chest was clear, heart sounds were normal and there were no neurological abnormalities. Laboratory tests including FBC, LFT, U&E and Creatinine were normal. Her MRI scan was not of a good quality due to limitation of resources and technology at place of her diagnosis, but it showed an 11.4 x 11.8cm solid, well-defined mass arising from pelvis and extending up to the umbilicus. The mass showed intermediate low signals on T1 and hyper intense signals on T2 images (Fig. 1). Mid line surgical exploration was undertaken which showed a large, solid, retroperitoneal mass arising from sacral nerves within the pelvis. Mass was lying in front of great vessels, overlapping the confluence of common iliac vessels. The left ureter was displaced laterally while the right ureter was lying over the mass. The mass was excised completely. Post operative course was uneventful and patient was discharged home on the fifth post operative day.
Figure1: MRI showing showing a large soft tissue mass.
Macroscopically, the specimen was a 13x13x5cm rounded well-encapsulated mass (Fig. 2). Upon sectioning in vitro, mass was seen to be solid, whorled and grey white. Microscopically, groups and singly scattered ganglion cells were seen with surrounding neural tissue. There was no evidence of atypia, mitosis or necrosis. Features were suggestive of a ganglioneuroma. (Figure 3) The patient was well at two months follow up and required no further treatment.
Figure2: Photograph of the resected specimen shows a well-encapsulated ovoid mass.
Neuroblastoma, ganglioneuroblastoma and ganglioneuromas are tumours of sympathetic nervous system that arise from the neural crest cells.1 These tumours differ only in their progressive degree of cellular and extracellular maturity, with ganglioneuroma being the most mature hence well differentiated and neuroblastoma being the least.2 Ganglioneuroma are rare, benign and slow growing. They may occur spontaneously or as a down grading from therapy for Neuroblastoma with either chemotherapy or radiation.3 International Neuroblastoma Pathology Classification (INPC) has been devised after studying 552 such tumours. Out of 300 with favourable prognosis three groups were identified as; ganglioneuroma maturing (GN-M), ganglioneuroblastoma intermixed (GNB-I) and ganglioneuroblastoma nodular with favourable subset (GNB-N-FS). These are resectable in 91% cases in one or more surgical sessions. In contrast, the remaining 252 tumours had unfavourable prognosis and were called ganglioneuroblastoma nodular unfavourable subset (GNB-N-US). This group was not amenable to surgical resection and usually already had metastasis at the time of presentation.4
Ganglioneuromas although are mostly sporadic, may be associated with Neurofibromatosis (Von Recklinghausens Disease) and Multiple Endocrine Neoplasia type II (MEN).1 Ganglioneuroma usually presents before the second decade and rarely after the sixth.2 The median age at diagnosis has been reported to be approximately 7 years. There is a slight female preponderance.5 The common locations are the posterior mediastinum, and the retroperitoneal space. Retroperitoneal pelvic location is very rare and only few case histories have been reported.1
Although retroperitoneal ganglioneuromas are usually asymptomatic, some patients may get compression symptoms, diarrhea, hypertension, virilization and myasthenia gravis owing to release of certain peptides.1 Radiological examination may localize the lesion. MRI may show low intensity on T1-weighted images and heterogenous hyper intensity on T2-weighted images with gradual increasing enhancement on dynamic images.6
Surgical excision is sufficient for treatment of ganglioneuromas. Chemotherapy or radiotherapy has no role in the treatment. Even with an incomplete excision, close follow up alone may be adequate. If any progression of the tumour is seen then repeat laparotomy may be indicated.2
Conclusion:
Although pelvic ganglioneuroma is a very rare lesion, it should be considered in the differential diagnosis of any abdomeno-pelvic mass. As it is a slow growing tumour, gross total surgical removal with preservation of organ function is a feasible surgical option.
Majority of pancreatic tumours are of primary pancreatic origin. Nevertheless a multitude of extra pancreatic cancers can metastasize to the pancreas and may present a diagnostic and management dilemma. Our case demonstrates such a problem in a patient with a pancreatic lesion.
Case report
A 82 year old man was referred to our hospital with computed tomogram (CT) scan showing a hypodense lesion in the pancreas. He had an anterior resection done 5 years prior for a Duke’s B (pT3N0M0) colon cancer. He did not receive any post-operative chemotherapy or radiotherapy. Carcinoembryonic antigen (CEA) levels was normal. He underwent an MRI scan (Figure 1) of his abdomen which reported a 2.8cm ring enhancing lesion in the tail of pancreas. At endoscopic ultrasound (EUS) a 2 x 2 cm well circumscribed mass was demonstrated in the tail of the pancreas close to the splenic artery but, not involving the vessel. Fine needle aspiration (FNA) of the lesion demonstrated a poorly differentiated mucin secreting adenocarcinoma. Immuno-histochemical staining was strongly positive for CK 20 but, CK 7 was only weak focally positive (Figure 2) thus, suggesting metastasis to the pancreas from a colonic primary as opposed to a primary pancreatic malignancy.
The patient was given an option to undergo subtotal pancreatectomy or consider palliative chemotherapy. The patient chose neither and was discharged home with input from the Macmillan team.
Figure 1: MRI after gadolinium showing a ring-enhancing lesion in the tail of pancreas.
Figure 2: (a) Fine needle aspirate on liquid based cytology (x 400) shows irregular distribution of cells with nuclear palisading and pleomorphism. Immunocytochemistry performed on cytology smear shows (b) strong positivity for CK 20 (c) negative for CK7 and (d) focal positivity for CA19.9.
Discussion:
The pancreas is an uncommon site of metastasis from other primary cancers.1 Most of the space occupying lesions seen in the pancreas on imaging are of primary pancreatic origin.1, 2 Adsay, et al 2 performed analyses on surgical and autopsy database in 2004 and found that amongst a total of 4955 adult autopsies and 973 pancreatic specimens at surgery; the prevalence of different metastatic tumours to the pancreas was only 1.6% of all examined autopsy cases and 3.9% of pancreatic resections.
A study from Japan found that the commonest primary malignancies to metastasize to the pancreas were from the stomach, lung and bile duct in that order.3 Other primary tumours that have been reported to metastasize to the pancreas include renal cell carcinoma, lung, breast, small bowel, colon, rectum and melanoma.4, 5 Several mechanisms for development of pancreatic metastases (particularly from colorectal cancer) have been described: transfer via the lymphatic system, metastases from peritoneal carcinomatosis, and/or transfer via the haematogenous system. 6 Direct invasion of the pancreas by the primary tumour was also noted to be a method of spread from bile duct and gastric malignancies.3
CT scan is often unhelpful in differentiating primary from secondary pancreatic lesions. Pancreatic metastasis can present as solid or cystic structures, hypodense or hyper dense lesions.7, 8 A series by Klein, et al in which the CT features of pancreatic tumours are described suggested that multiplicity of tumours and/or hypervascularity were characteristic of secondary pancreatic tumours.9 A recent study has suggested that Positron Emission Tomogram (PET) is a more sensitive investigative tool than CT in detecting metastatic colorectal cancer.10 Most patients (as in our unit) usually have EUS guided FNA or biopsy to arrive at a diagnosis.
The differential diagnosis of primary pancreatic cancer versus metastasis from other carcinomas may be difficult using common histopathological techniques.11 Immuno-histochemical staining is often helpful in differentiating primary from secondary pancreatic tumours. Sometimes staining by a combination of different antibodies helps to reach a diagnosis. In a survey of 435 cases, the expression of CK 7 was positive in 92% of pancreatic cancers but in only 5% of colon cancers. On the other hand CK 20 was positive in 100% of colon cancers and in only 62% of pancreatic cancers.12 Furthermore, CD X2 is frequently expressed in colorectal carcinoma but, rarely in pancreatic ductal adenocarcinoma.13
The choice between conservative chemotherapy and resection for solitary pancreatic metastasis from colorectal cancer is still undecided. The natural history of untreated patients with pancreatic metastasis from cancer of the colon or rectum is unknown and thus it is impossible to compare the survival rate of resected and unresected patients treated with chemotherapy.14 Researchers from John Hopkins have reported only 4 colon metastasis to the pancreas (0.6%) among 650 pancreatico-duodenectomy procedures performed in their institution from 1990 to 1996.15 Experience from an Italian centre14 published that metastasis to the pancreas was the indication for surgery in a total of 18 out of 546 pancreatic resections (3.2%) performed over 27 years and colorectal cancer was the primary tumour in 50% of those cases. The median survival time was 16.5 months (range 8 – 105 months) with no peri-operative mortality being reported. In another study, all symptomatic (pain or jaundice) patients experienced complete relief of symptoms after surgery and no one experienced obstructive jaundice or abdominal pain until tumour recurrence.16
Oncologists may argue that chemotherapy can offer the same results as pancreatic resection but with less morbidity. Unfortunately, there is paucity of data in medical literature on comparisons of outcomes associated with surgical and chemotherapeutic treatment. We agree with Sperti et al 14 that resection of pancreatic metastasis from colorectal cancer is a palliative procedure with long-term survival being an exceptional event.
Conclusion:
Our case demonstrates that differential diagnoses for pancreatic masses should always include metastasis to the pancreas from other tumours particularly, when there is a history of previous or concurrent non-pancreatic malignancy. When disseminated malignancy is not present an aggressive surgical approach may offer successful palliation of symptoms and have a role in the multidisciplinary management of metastatic malignancy.
A 67 year old Caucasian male presented to our institution with a one day history of uncontrollable movements. The patient was being evaluated by a psychiatrist, neurologist and a neuro-opthalmologist for a three month history of severe anxiety, gait instability and palinopsia, respectively. The patient had progressively worsened over the prior two weeks and at the time of presentation reported visual hallucinations with increased confusion. His involuntary movements escalated to the point where it appeared that he was having seizures.
His medical history was significant for gouty arthritis, hypertension, and major depression. His surgical history was notable for an open reduction and internal fixation of his left hip 6 years prior. There was no history of any blood or blood product transfusion. He was an insurance executive and did not have significant occupational exposures. His social and family history was unremarkable. His medications upon arrival included captopril, atenolol, bupropion, lamotrigine, clonazepam, folic acid, and ibuprofen.
On admission he was arousable, well nourished, afebrile, haemodynamically stable but disorientated. His cardiopulmonary, abdominal and integumentary examinations were unremarkable. Neurological examination was significant for bilateral symmetric hyperreflexia, diffuse cogwheel rigidity without a resting or postural tremor, and multi-focal dysrhythmic generalised myoclonus. No neck tenderness or nuchal rigidity was noted. A CT of the head without contrast in the causality department was negative for haemorrhage or any acute intracranial pathology.
His initial assessment showed confusion, hallucinations, myoclonus with medication induced delirium. Lewy body dementia, occipital lobe epilepsy, peduncular hallucinosis and prion disease were all considered in the differential diagnosis. On admission, laboratory data including a CBC, CMP, serum ammonia level, cardiac enzymes, urinalysis, and coagulation profile were unremarkable. A toxicology screen for illicit drugs and heavy metals was negative. The initial lumbar spinal fluid (CSF) analysis was only notable for mild proteinorachia of 57 mg/dL. Gram stain, India-ink stain, acid-fast bacilli (AFB), bacterial & fungal cultures, as well as PCR for viral nucleic acid (herpes, Varicella-Zoster, Epstein-Barr virus, arboviruses, and cytomegalovirus virus) were all negative. MRI with contrast was remarkable only for periventricular ischemic changes consistent with small vessel disease. The patient’s bupropion and lamotrigine were discontinued upon admission, and his clonazepam was increased with resolution of the myoclonus after 24 hours.
The admission EEG showed diffuse slowing with no epileptiform discharges or triphasic waves. Due to progressive neurologic deterioration, he was followed with serial electroencephalograms. On hospital day 5, he became unresponsive and the subsequent EEG revealed non-convulsive status epilepticus (NCSE). Temporary resolution of his seizures was achieved with lorazepam and pentobarbital infusions. After 3 days of almost complete suppression, the pentobarbital was discontinued without NCSE recurrence. On hospital day 15 the EEG again displayed NCSE. A ketamine drip was added to his drug regimen with only brief improvement. Pentobarbital had been restarted and progressively titrated up to the maximal dose without achieving burst suppression. Despite being on the maximal dose of pentobarbital, ketamine, valproic acid, levetiracetam, and topiramate he continued to display NCSE (Figure 1A).
At this point (hospital day 16), therapeutic hypothermia was initiated and continued for 48 hours. The patient’s core body temperature was maintained between 32-33 °C followed by slow rewarming to normothermia over the following 48 hours. Near complete suppression of epileptiform activity was observed on the EEG (Figure 1B). Ketamine and pentobarbital were successfully weaned off during the following days and phenobarbital was introduced without recurrence of NCSE.
Figure 1A. Electroencephalogram from hospital day 15. Refractory non-convulsive status epilepticus while on ketamine, levetiracetam, valproic acid, topiramate and pentobarbital.
Figure 1B. Electroencephalogram from hospital day 16, after the initiation of therapeutic hypothermia. Suppression of epileptiform activity is observed after treatment with therapeutic hypothermia; ECG artifact persisting.
Figure 1C. Electroencephalogram from hospital day 29, 13 days after treatment with therapeutic hypothermia, illustrating generalised periodic sharp wave discharges with lack of background activity. Occasional triphasic waves are noted consistent with Creutzfeldt-Jakob encephalopathy.
Figure 2. RepeatMRI of the brain on hospital day 21 illustrates asymmetric basal ganglia hyperintensities on diffusion weighted sequences, which are often observed in CJD.
Figure 3. H & E stain of the cerebral cortex with low and high magnification (A & B). Coarse and fine vacuolization with spongiosis (arrows) are demonstrated on H & E and silver stain, respectively (C & D).
The patient had a repeat MRI which showed asymmetric basal ganglia hyper intensity on diffusion weighted imaging sequence consistent with CJD 3 (Figure 2)3. The results of CSF analysis for protein 14-3-3, neuron-specific enolase, and tau protein became available on hospital day 22. Despite controlling the NCSE, the patient remained unresponsive over the course of the following weeks. The EEG pattern changed to generalised periodic sharp waves, 1-2 per second with occasional triphasic waves, and a lack of background activity (Figure 1C). After fully reviewing the results with the family, an open brain biopsy was performed in effort to verify the diagnosis. The biopsy confirmed the diagnosis of spongiform encephalopathy (Figure 3). In light of the findings, withdrawal of care was initiated upon the family’s request and the patient expired on hospital day 42. The patient’s estimated symptomatic clinical course was approximately four and one-half months.
DISCUSSION
Creutzfeldt-Jakob disease is the archetype of prion mediated neurodegenerative disorders. There are 4 types of CJD; sporadic, familial, iatrogenic and variant4. The sporadic type accounts for 85 per cent of all cases of CJD4. The diagnosis of CJD and transmissible spongiform encephalopathy (TSE) can be elusive. The World Health Organisation’s diagnostic criteria for CJD require at least one of the following: (1) Neuropathological confirmation, (2) confirmation of protease-resistant prion protein (PrP) via immunohistochemistry or Western blot, or (3) presence of scrapie-associated fibrils4. However, newer and less invasive means for diagnosis have been explored in recent literature. CSF analysis for protein 14-3-3, tau protein, S100B protein and neuron-specific enolase have demonstrated sensitivities of 93 per cent, 89 per cent, 87 per cent and 78 per cent, respectively5. In addition, the use of MRI fluid-attenuated inversion recovery (FLAIR) and diffusion weighted imaging (DWI) techniques have yielded sensitivities of 91-92 per cent and specificities of 94 -95 per cent respectively, especially when utilised early in the disease state6. In our case, the initial MRI was unremarkable and only the repeated MRI, performed three weeks after admission, revealed basal ganglia signal intensities consistent with CJD.
One of the most studied and well characterised tools used to support the diagnosis of CJD is the EEG. The typical pattern observed in the early stages of CJD is frontal intermittent rhythmic delta activities (FIRDA). As the disease progresses, characteristic periodic sharp wave complexes (PSWC) can be observed, usually 8 to 12 weeks after the onset of symptoms7. However, the reported sensitivity of EEG is relatively low, ranging from 22 to 73 per cent, and is largely dependent of the subtype of CJD8. In our case, the patient presented with NCSE, which is an uncommon presentation of an uncommon disease. In a retrospective review of 1,384 patients with probable or definite CJD, only 0.007 per cent or 10 patients presented with NCSE2. Our patient did not demonstrate EEG findings consistent with CJD until late in his hospital course. Hence, CJD must be considered as a diagnosis in a patient who presents with refractory non-convulsive status epilepticus without overlooking the more common causes9.
The last important observation is the potential utility of therapeutic hypothermia in patients with refractory NCSE. Therapeutic hypothermia has long been known to suppress epileptiform discharge10,11. However, the safety and efficacy have not been broadly studied in human subjects. Corry and colleagues conducted a small study examining the effects of therapeutic hypothermia on 4 patients with refractory status epilepticus. The results were promising in that therapeutic hypothermia was successful in aborting seizure activity in all 4 patients and effectively suppressed seizure activity in 2 of the 4 patients after re-warming12. We were able to observe similar result in achieving temporary resolution of NCSE with therapeutic hypothermia in combination with antiepileptic medication in our patient.
Malaria is caused by obligate intra-erythrocytic protozoa of the genus Plasmodium. Humans can be infected with one (or more) of the following five species: P. falciparum, P. vivax, P. ovale, and P. malariae and P. knowlesi. Plasmodia are transmitted by the bite of an infected female Anopheles mosquito and these patients commonly present with fever, headache, fatigue and musculoskeletal symptoms.
Diagnosis is made by demonstration of the parasite in peripheral blood smear. The thick and thin smears are prepared for identification of malarial parasite and genotype respectively. Rapid diagnosis of malaria can be done by fluorescence microscopy with light microscope and interference filter or by polymerase chain reaction.
We report a complicated case of P. ovale malaria without fever associated with Hepatitis B virus infection, pre-excitation (WPW pattern), and secondary adrenal insufficiency.
Case Report:
A 23 year old African American man presented to the emergency department with headache and dizziness for one week. He had 8/10 throbbing headaches associated with dizziness, nausea and ringing sensation in the ears and also complained of sweating but denied any fever. He had loose, watery bowel movements 3 times a day for a few days and had vomited once 5 days ago. He denied any past medical history or family history. He was a chronic smoker and smoked 1PPD for 8 years and denied alcohol or drug use. He had travelled to Africa 9 months before presentation and had stayed in Senegal for 1 month though he did not have any illnesses during or after returning from Africa.
On examination: T: 97.6, HR: 115/min, BP: 105/50, no orthostasis, SPO2: 100% in room air and RR: 18/min. Head, neck and throat examinations were normal and respiratory and cardiovascular system examinations were unremarkable except for tachycardia. Abdominal examination revealed no organomegaly and his CNS examination was unremarkable.
Laboratory examination revealed: WBC: 6.4, Hb: 14.4 and Hct: 41.3, Platelets: 43, N: 83.2, L: 7.4, M: 9.3, B: 0.1. His serum chemistry was normal except for a creatinine of 1.3 (BUN 14) and albumin of 2.6 (total protein 5.7). A pre-excitation (WPW Pattern) was seen on ECG and head CT and Chest X-ray were normal.
He was admitted to the telemetry unit to monitor for arrhythmia. Peripheral blood smear (PBS) was sent because of thrombocytopenia and mild renal failure and revealed malarial parasites later identified as P. ovale (Pic. 1 and 2).
He was treated with Malarone; yet after 2 days of treatment, he was still complaining of headache, nausea and dizziness. There were no meningeal signs. His blood pressure readings were low (95/53) and he was orthostatic. His ECG showed sinus tachycardia and did not reveal any arrhythmias or QTc prolongation. His morning serum cortisol was 6.20 and subsequent cosyntropin stimulation test revealed a serum cortisol of 13.40 at one hour after injection. His Baseline ACTH was<1.1 suggesting a secondary adrenal insufficiency. His IGF-1, TSH, FT4, FSH, LH were all within normal limits. His bleeding and coagulation parameters were normal, CD4 was 634(CD4/CD8: 1.46) and rapid oral test for HIV was negative. His Hepatitis B profile was as follows: HBsAg: positive, HBV Core IgM: negative, HBV core IgG: positive, HBeAg: negative, HBeAb: positive, HBV DNA: 1000 copies/ml, Log10 HBV DNA: 3000 copies/ml.
His Blood cultures were negative, his G6PD levels and hemoglobin electrophoresis were normal, haptoglobin was<15 and LDH was 326. MRI of the brain was unremarkable. The abdominal sonogram revealed a normal echo pattern of the liver and spleen and spleen size was 12 cm. The secondary adrenal insufficiency was treated with dexamethasone resulting in gradual improvement of his nausea, vomiting and headache. Furthermore the platelet count improved to 309. Primaquine was prescribed to complete the course of malaria treatment and he was discharged home following 8 days of hospitalization. Unfortunately he did not return for follow up.
Discussion:
Malaria continues to be a major health problem worldwide. In 2007 the CDC received reports of 1,505 cases of malaria among person in the United States. 326 cases were reported from New York with all but one of these cases being acquired outside of the United States1.
While Plasmodia are primarily transmitted through the bite of an infected female Anopheles mosquito, infections can also occur through exposure to infected blood products (transfusion malaria) or by congenital transmission. In industrialized countries most cases of malaria occur among travellers, immigrants, or military personnel returning from areas endemic for malaria (imported malaria). Exceptionally, local transmission through mosquitoes occurs (indigenous malaria). For non-falciparum malaria the incubation period is usually longer (median 15–16 days) and both P. Vivax and P. Ovale malaria may relapse months or years after exposure due to the presence of hypnozoites in the liver of which the longest reported incubation period for P. vivax being 30 years2.
Malaria without fever has been reported in cases of Plasmodium falciparum malaria in non- immune people3. Hepatitis B infection associated with asymptomatic malaria has been reported in the Brazilian Amazon4. This study was done in P. falciparum and P. vivax infected person with HBV co-infection though not in the P. ovale group. HBV infection leads to increased IFN-gamma levels5,6 which are important for plasmodium clearance in the liver7, in addition to its early importance for malarial clinical immunity8. High levels of IFN gamma, IL6 and TNF alpha are detectable in the blood of malaria patients and in the spleen and liver in the rodents’ model of malaria9,10. These inflammatory cytokines are known to suppress HBV replication in HBV transgenic mice9. This might explain the low levels of HBV viremia in our patient although human studies are required to confirm this finding.
The hypothalamic-pituitary- adrenocortical axis suppression and primary and secondary adrenal insufficiency has been reported in severe falciparum malaria10. In our case, the patient did not have any features to characterize severe malaria, and parasitaemia was <5%. Further, the MRI did not reveal any secondary cause for adrenal insufficiency. This might indicate that patients with malaria are more prone for hypothalamo-pituitary adrenocortical axis dysregulation yet further studies are required to prove this phenomenon in patients without severe malaria.
Cardiac complications after malaria have rarely been reported. In our patient pre-excitation on ECG disappeared after starting antimalarial treatment. Whether WPW pattern and its subsequent disappearance was incidental or caused by malarial infection that improved with treatment could not be determined. Lengthening of the QTc and severe cardiac arrhythmia has been observed, particularly after treatment with halofantrine for chloroquine resistant Plasmodium falciparum malaria11. Post-infectious myocarditis can be associated with cardiac events especially in combination with viral infections12. A case of likely acute coronary syndrome and possible myocarditis was reported after experimental human malaria infection13. To date, except for cardiac arrhythmias that developed after treatment with halofantrine and quinolines, no other arrhythmias has been reported in patients with malaria before treatment.
Transient thrombocytopenia is very common in uncomplicated malaria in semi -immune adults14. A person with a platelet count <150 × 109/l is 4 times more likely to have asymptomatic malarial infection than one with a count ≥150 × 109/l15. In an observational study among 131 patients, patients with involvement of more than one organ system was found to have a lower mean platelet count compared to those with single organ involvement16.
Conclusions:
Our case highlights the need for further studies to understand the multi-organ involvement in patients without severe malaria as well as early recognition of potential complications to prevent mortality and morbidity in this subgroup of patients.
A 69 year old male with hypertension, body mass index 24 kg/m2, neck circumference 16 inches, and moderate COPD, on home oxygen, presented to his pulmonary clinic appointment with worsening complaints of fatigue, leg cramps, and intermittent shortness of breath with chest discomfort. A remote, questionable history of syncope five to ten years ago was elicited. His vital signs were: temperature 98.80F, blood pressure 119/76 mmHg, pulse 92/min and regular, and respirations 20/min. Physical exam was significant for crowded oropharynx with a Mallampati score of four, distant breath sounds with a prolonged expiratory phase on lung exam with a normal cardiac exam. Laboratory investigation showed normal complete blood counts, haemoglobin 15 g/dL, and normal chemistries. Compared to his previous studies, a pulmonary function study showed stable parameters with a FEV1 1.47 L (69%), FVC/FEV1 ratio 0.44 (62%), and a DLCO/alveolar volume ratio of 2.12 (49%). A room air arterial blood gas revealed pH 7.41, PCO2 44 mmHg, and PO2 61 mmHg, with 92% oxygen saturation. A six minute treadmill exercise test performed to assess the need for supplemental oxygen showed that he required supplemental oxygen at 1L/min via nasal cannula to eliminate hypoxemia during exercise. His chest radiograph was significant for hyperinflation and prominence of interstitial markings. A high resolution computed tomography of the chest demonstrated severe centrilobular and panacinar emphysema only. A baseline electrocardiogram (EKG) showed normal sinus rhythm with an old anterior wall infarct (Figure 1). Echocardiography of the heart revealed a normal left ventricle with an ejection fraction of 65%. Right ventricular systolic function was normal although elevated mean pulmonary arterial pressure of 55 mmHg was noted. A diagnostic polysomnogram performed for evaluation of daytime fatigue and snoring at night revealed mild OSA with an AHI of 6/hr. with sleep time spent with oxygen saturation below 90% (T-90%) of 19%. The EKG showed normal sinus rhythm. A full overnight polysomnogram for continuous positive airway pressure (CPAP) titration performed for treatment of sleep disordered breathing was sub-optimal, however it demonstrated an apnoea–hypopnea index (AHI) of 28 during REM (rapid eye movement) sleep, and a T-90% of 93%. The associated electrocardiogram showed Wenckebach second degree AV heart block during REM sleep usually near the nadir of oxygen desaturation. On a repeat positive airway pressure titration study, therapy with Bilevel pressures (BPAP) of 18/14 cmH20 corrected the AHI and nocturnal hypoxemia to within normal limits during Non REM (NREM) and REM sleep. His electrocardiogram remained in normal sinus rhythm .A twenty-four hour cardiac holter monitor revealed baseline sinus rhythm and confirmed the presence of second degree AV block of the Wenckebach type. A one month cardiac event recording showed normal sinus rhythm with frequent episodes of second degree AV block. These varied from Type I progressing to Type II with a 2:1 and 3:1 AV block, during sleep. Progression to complete heart block was noted with the longest pause lasting 3.9 seconds during sleep. The patient underwent an electrophysiology study with placement of a dual chamber pacemaker. He was initiated on BIPAP therapy. Subsequently, the patient was seen in clinic with improvements in his intermittent episodes of shortness of breath, fatigue, and daytime sleepiness.
Figure 1- Patient’s baseline EKG, normal sinus rhythm. Figure 2 -Progression to Mobitz Type II block 5:07 am. Figure 3 and 4- Sinus pauses, longest interval 11:07 pm 3.9 seconds (Figure 4).
Discussion
In healthy individuals, especially athletes, bradycardia, Mobitz I AV block, and sinus pauses up to 2 seconds are common during sleep and require no intervention5. Cardiac rhythm is controlled primarily by autonomic tone. NREM sleep is accompanied by an increase in parasympathetic, and a decrease in sympathetic, tone. REM sleep is associated with decreased parasympathetic tone and variable sympathetic tone. Bradyarrhythmias in patients with OSA are related to the apnoeic episodes and over 80% are found during REM sleep. During these periods of low oxygen supply, increased vagal activity to the heart resulting in bradyarrhythmias may actually be cardioprotective by decreasing myocardial oxygen demand. This may be important in patients with underlying coronary heart disease.
Some studies have found that Mobitz I AV block may not be benign. Shaw6 et al studied 147 patients with isolated chronic Mobitz I AV block. They inserted pacemakers in 90 patients, 74 patients were symptomatic and 16 patients received a pacemaker for prophylaxis. Outcome data included five-year survival, deterioration of conduction to higher degree AV block, and new onset of various forms of symptomatic bradycardia. They concluded that survival was higher in the paced groups and that risk factors for poor outcomes in patients with Mobitz I included age greater than 45 years old, symptomatic bradycardia, organic heart disease, and the presence of a bundle branch block on EKG.
The Sleep Heart Health Study7 found a higher prevalence of first and second-degree heart block among subjects with sleep-disordered breathing (SDB) than in those without (1.8% vs. 0.3% and 2.2 vs. 0.9%, respectively). Gami et al8observed thatupon review of 112 Minnesota residents who hadundergone diagnostic polysomnography and subsequentlydied suddenly from a cardiac cause, sudden death occurred between the hours of midnight and 6:00 AM in 46% of those with OSA, as compared with 21% of those without OSA. In a study of twenty-three patients with moderate to severe OSA who were each implanted with an insertable loop recorder, about 50% were observed to have frequent episodes of bradycardia and long pauses (complete heart block or sinus arrest) during sleep9. These events showed significant night-to-night intra individual variability and their incidence was under-estimated, only 13%, by conventional short-term EKG Holter recordings.
Physiologic factors predisposing patients with OSA to arrhythmias include alterations in sympathetic and parasympathetic nervous system activity, acidosis, apnoea’s, and arousal2, 10, 11. Some patients with OSA may have an accentuation of the ‘Diving Reflex’. This protective reflex consists of hypoxemia-induced sympathetic augmentation to muscles and vascular beds associated with increased cardiac vagal activity which results in increased brain perfusion, bradycardia and decreased cardiac oxygen demand. In patients with cardiac ischemia, poor lung function (i.e. COPD), or both, it may be difficult to differentiate between these protective OSA-associated Bradyarrhythmias and those which may lead to sudden death. It has been well established that patients with COPD are at higher risk for cardiovascular morbidity12 and arrythmias13. Fletcher14 and colleagues reported that the effects of oxygen supplementation on AHI, hypercapnea and supraventricular arrhythmias in patients with COPD and OSA were variable. Out of twenty obese men with COPD studied, in most patients oxygen eliminated the bradycardia observed during obstructive apnoea’s and eliminated AV block in two patients. In some patients supplemental oxygen worsened end-apnoea respiratory acidosis however this did not increase ventricular arrhythmias.
CPAP therapy has been demonstrated to significantly reduce sleep–related Bradyarrhythmias, sinus pauses, and the increased risk for cardiac death 9, 15. Despite this, in certain situations placement of a pacemaker may be required. These include persistent life-threatening arrhythmias present in patients with severe OSAS on CPAP, arrhythmias in patients who are non-compliant with CPAP, and in patients who may have persistent sympathovagal imbalance and hemodynamic fluctuations resulting in daytime bradyarrhythmias16.
Our case is interesting since it highlights the importance of recognizing the association between OSA, COPD, and life-threatening cardiac arrhythmias. Primary care providers should note the possible association of OSA-associated bradyarrhythmias with life-threatening Type II bradyarrhythmias and pauses. Since bradyarrhythmias related to OSA are relieved by CPAP, one option would be to treat with CPAP and observe for the elimination of these arrhythmias using a 24hour holter or event recorder17. Compliance with CPAP is variable and if life-threatening bradycardia is present, placement of a permanent pacemaker may be preferred18.
Our patient is unusual because most studies showing a correlation with the severity of OSA and magnitude of bradycardia have included overweight patients without COPD19. This patient’s electrocardiogram revealed a Type II AV block at 5am (Figure 2). This is within the overnight time frame where patients with OSA have been observed to have an increased incidence of sudden death. Figures 3 and 4 show significant sinus pauses. In selected cases where patients have significant co-morbidities (i.e. severe COPD with OSA), in addition to treatment with positive airway pressure, electrophysiological investigation with placement of a permanent pacemaker may be warranted.
Although one cell type may predominate, teratomas usually comprise of tissue from all three embryonic germ layers1. Generally arising from the gonads, they may be found in extra-gonadal sites such as sacro-coccygeal region, mediastinum, neck and retroperitoneum.2 Here we report a case of retroperitoneal teratoma in an adult with successful surgical treatment. Its clinical presentation, diagnosis and treatment are reviewed.
Case Report:
A woman aged 28 years presented with pain in the right hypochondrium of one year duration. There was no associated bowel or urinary symptom. Examination showed minimal fullness in the right hypochondrium. Routine blood tests and urinalysis were within normal limits. A plain abdominal radiograph showed calcification in the right side of the abdomen (Fig. 1). Ultrasonography demonstrated 13.6 x 8.1 cm soft tissue mass in the retro-peritoneum between liver and the right kidney. It was heterogeneous, well circumscribed with sharply defined borders, and had some calcification and cystic areas. CT abdomen revealed a hypo-dense lesion between liver and the right kidney. It had fatty attenuation with internal hyper-dense areas representing calcification. (Fig. 2). Provisional diagnosis of a retroperitoneal teratoma was made and an open exploration was performed with a right sub-costal incision. There was a large cystic mass behind the ascending colon, duodenum and the pancreas. It was located in the retroperitoneal compartment. There were dense, fibrous adhesions of the mass with aorta but entire cystic mass was excised successfully.
Post operatively this tumor mass measuring 5 x 5 cm was excised in vitro and found to be filled with yellowish creamy material containing hair, sebum and bony tissue. Microscopically it was confirmed to be a cystic teratoma with no malignancy. Stratified squamous epithelium with sebaceous and sweat glands, hair shafts, calcification, few bony spicules and bone marrow elements were all demonstrated. (Fig. 3). The post operative course was uneventful and she was well at the 2 months follow up.
Figure 1. Plain abdominal radiograph showing radio-opaque shadow (arrow heads) in the right upper abdomen.
Figure 2: Computed Tomography showing an encapsulated mass that contains multiple tissue elements including fat and areas of calcification.
Figure 3: Microscopic examination of the tumor showing squamous epithelium (SE), hair shaft (HS), sebaceous glands (SBG)
Discussion:
Teratomas are congenital tumours arising from pluri-potential embryonic cells and therefore have several recognizable somatic tissues3, Teratomas are usually localized to the ovaries, testis, anterior mediastinum or the retro-peritoneal area in descending order of frequency.4 Teratomas constitute less than 10% of all primary retroperitoneal tumours and hence are relatively uncommon5. Furthermore, retroperitoneal teratomas occur mainly in children and have been very rarely described in the adults. Half of these cases present in children less than 10 years of age and only a fifth of them present after 30 years of age. Retroperitoneal teratomas are often located near the upper pole of the kidney with preponderance on the left. The case described here is therefore unusual in that it was a primary retroperitoneal teratoma in an adult, on the right side and with adhesions to the aorta.
Retroperitoneal teratomas are seen in females twice as commonly than males. Teratomas are usually benign if they are cystic and contain sebum or mature tissue. They are more likely to be malignant if they are solid and have immature embryonic tissue like fat, cartilage, fibrous and bony elements.6 In these regards our case is similar to other described cases as our patient is also female and as her teratoma was cystic, it showed lack of malignancy.
Teratomas are usually asymptomatic as the retroperitoneal space is extensive enough to allow for their free growth. When compression of the surrounding structure occurs, patients may get compression symptoms.The diagnosis of a retroperitoneal teratoma cannot be made on clinical grounds alone. Ultrasound and computed tomography are important in its diagnosis and may show the presence of calcification, teeth or fat. Calcification on the rim of tumour or inside the tumour is seen in 53-62% of teratomas and although radiologically three quarters of patients with a benign teratoma may have calcification within it, a quarter of malignant cases may also demonstrate calcification. Computed tomography is better than Ultrasonography in defining the extent and spread of teratoma to the surrounding organs.7
The prognosis is excellent for benign retroperitoneal teratoma if complete resection can be accomplished.
Even though it is commonly seen in Graves' disease, TPP is not related to the etiology, severity, and duration of thyrotoxicosis. 1
The pathogenesis of hypokalaemic periodic paralysis in certain populations with thyrotoxicosis is unclear. Transcellular distribution of potassium is maintained by the Na+/K+–ATPase activity in the cell membrane, and it is mainly influenced by the action of insulin and beta-adrenergic catecholamines.2 Hypokalemia in TPP results from an intracellular shift of potassium and not total body depletion. It has been shown that the Na+/K+–ATPase activity in platelets and muscles is significantly higher in patients with TPP.3 Hyperthyroidism may result in a hyperadrenergic state, which may lead to the activation of the Na+/K+–ATPase pump and result in cellular uptake of potassium.2, 4, 5 Thyroid hormones may also directly stimulate Na+/K+– ATPase activity and increase the number and sensitivity of beta receptors.2, 6 Patients with TPP have been found to have hyperinsulinemia during episodes of paralysis. This may explain the attacks after high-carbohydrate meals.7
CASE REPORT:
A 19 year old male patient presented to our emergency room with sudden onset weakness of lower limbs. He was not able to stand or walk. Power of 0/5 in both lower limbs and 3/5 in upper limbs was noticed on examination. Routine investigations revealed to have severe hypokalemia with a serum potassium of 1.6 meq/l (normal range 3.5-5.0 meq/l), a serum phosphorus level of 3.4 mg/dl (normal range 3-4.5 mg/dl) and mild hypomagnesemia with serum magnesium level of 1.5mg/dl (normal range 1.8-3.0 mg/dl). ECG showed hypokalemic changes with prolonged PR interval, increased P-wave amplitude and widened QRS complexes. He was managed on intravenous as well oral potassium and history revealed weight loss, increased appetite and tremors from past 4 months. He had a multinodular goiter and radioactive iodine uptake scan (Iodine 131) showed a toxic nodule (Toxic nodule shows increased iodine uptake while the rest of the gland is suppressed) with no exophthalmos, sensory or cranial nerve deficits. Thyroid function tests revealed thyrotoxicosis with free T4 of 4.3ng/dl (normal range 0.8-1.8ng/dl), T3 of 279 ng/dl (normal range = 60 - 181 ng/dl) and a TSH level of <0.15milliunits/L (normal range = 0.3 - 4 milliunits/L). He was managed on intravenous potassium & propanolol. The patient showed dramatic improvement of his symptoms. The patient was discharged home on carbamazole with the diagnosis of TPP secondary to toxic nodular goiter.
In this case there was a significant family history as one of his elder brother had a sudden death (cause not known) and his mother was primary hypothyroid on levothyroxin replacement therapy.
DISCUSSION :
TPP is seen most commonly in Asian populations, with an incidence of approximately 2% in patients with thyrotoxicosis of any cause.1,8,9,10 The attacks of paralysis have a well-marked seasonal incidence, usually occurring during the warmer months.1 Pathogenesis of hypokalaemia has been explained by some authors to be due to an intracellular shift of body potassium, which is catecholamine mediated.11,12 Shizume and his group studied total exchangeable potassium which revealed that patients with thyrotoxic periodic paralysis were not significantly different from controls when the value was related to lean body mass.11 The paralytic symptoms and signs improve as the potassium returns from the intracellular space back into the extracellular space.13 The diurnal variation in potassium movement where there is nocturnal potassium influx into skeletal muscle would explain the tendency for thyrotoxic periodic paralysis to occur at night.14 Hypophosphataemia and hypomagnesaemia are also known to occur in association with thyrotoxic periodic paralysis.14,15,16,17,18 The correction of hypophosphataemia without phosphate administration supports the possibility of intracellular shift of phosphate.16 Electrocardiographic findings supportive of a diagnosis of TPP rather than sporadic or familial periodic paralysis are sinus tachycardia, elevated QRS voltage and first-degree AV block (sensitivity 97%, specificity 65%).20 In addition to ST-segment depression, T-wave flattening or inversion and the presence of U waves are typical of hypokalaemia.
The management is to deal with the acute attack as well as treatment of the underlying condition to prevent future attacks. Rapid administration of oral or intravenous potassium chloride can abort an attack and prevent cardiovascular and respiratory complications.4 A small dose of potassium is the treatment of choice for facilitating recovery and reducing rebound hyperkalaemia due to release of potassium and phosphate from the cells on recovery.1,2,3 Rebound hyperkalaemia occurred in approximately 40% of patients with TPP, especially if they received >90 mmol of potassium chloride within the first 24 hours.4 Another mode of treatment is to give propranolol, a nonselective b-blocker, which prevents the intracellular shift of potassium and phosphate by blunting the hyperadrenergic stimulation of Na+/K+–ATPase.20 Hence, initial therapy for stable TPP should include propranolol.21,22,23 The definitive therapy for TPP includes treatment of hyperthyroidism with antithyroid medications, surgical thyroidectomy, or radioiodine therapy.
The clinical features of early HAT are well defined, yet the features of delayed HAT are less clear. Delayed HAT is a rare complication of OLT that may present with biliary sepsis or remain asymptomatic. Sonography is extremely sensitive for the detection of HAT in symptomatic patients during the immediate postoperative period. However, the sensitivity of ultrasonography diminishes as the interval between transplantation and diagnosis of HAT increases due to collateral arterial flow. MRA is a useful adjunct in patients with indeterminate ultrasound exams and in those who have renal insufficiency or an allergy to iodinated contrast.
In the absence of hepatic failure, conservative treatment appears to be effective for patients with HAT but retransplantation may be necessary as a definitive treatment.
Case Presentation:
A 52 year old male with a history of whole graft OLT for primary sclerosing cholangitis presented with two days of fever, nausea, and mild abdominal discomfort.
One week prior to presentation, he was seen in the liver clinic for regular follow-up. At that time, he was totally asymptomatic and his laboratory workup including liver function tests were within normal range.
He has undergone OLT three years prior. At the time of transplant he required transfusion of 120 units of packed red blood cells, 60 units of fresh frozen plasma and 100 units of platelets due to extensive intraoperative bleeding secondary to chronic changes of pancreatitis and severe portal hypertension, but had an otherwise uneventful postoperative recovery.
On physical examination the temperature was 39C, heart rate was 125 beats per minute, respiratory rate was 22 bpm. Initial laboratory workup revealed a white blood cell count of 25,000/mm3, AST of 6230 U/L, ALT of 2450 U/L, total bilirubin of 11 mg/dL , BUN 55 mg/dL and Creatinine of 4.5 mg/dL. Lactate level was 5 mmol/L. Doppler ultrasonography revealed an extensive intrahepatic gas (Image 1A). Computed tomography of the abdomen and pelvis revealed extensive area of hepatic necrosis with abscess formation measuring 19x14 cm with extension of gas into the peripheral portal vein branches (Image 1B,C). Upon admission to the hospital, the patient required endotracheal intubation, mechanical ventilator support and aggressively fluid resuscitation. He was started on broad-spectrum antibiotics and a percutaneous drain was placed that drained dark, foul smelling fluid. Cultures from the blood and the drain grew Clostridium perfringens.
Magnetic resonance imaging (MRI), MRA revealed occlusion of the hepatic artery 2 cm from its origin and also evidence of collaterals (Image 2A,B).
Image 1: (Pannel A) Doppler ultrasonography reveal extensive intrahepatic gas. (Pannel B&C) Computed tomography of the abdomen and pelvis reveal an extensive area of hepatic necrosis with abscess formation measuring 19x14 cm with extension of gas into the peripheral portal vein branches.
Image 2: MRI & MRA reveal occlusion of the hepatic artery 2 cm from its origin and also evidence of collaterals.
Following drain placement, the patient’s clinical condition markedly improved with significant reduction of liver function test values. Retransplantation was considered but delayed in the setting of infection and significant clinical and laboratory testing improvement.
The patient was transferred to the medical floor in stable condition, and the drain was then removed.
A week later the patient developed low grade fevers and tachycardia. One day later he began to experience mild abdominal discomfort and high grade fevers. Repeat CT of the abdomen revealed worsening hepatic necrosis and formation of new abscesses. His clinical condition decompensated quickly thereafter requiring endotracheal intubation, mechanical ventilation and aggressive resuscitation. Percutaneous drain was placed and again, drained pus-like, foul-smelling material. His overall condition deteriorated, and he eventually expired a few days later.
Discussion:
Delayed (more than 4 weeks after transplantation) HAT is a rare complication of OLT with an estimated incidence of at around 2.8%1.
Risk factors associated with development of HAT include Roux-en-Y biliary reconstruction, cold ischaemia and operative time, the use of greater than 6 units of blood, the use of greater than 15 units of plasma, and the use of aortic conduits on arterial reconstruction during transplant surgery2.
Collateralization is more likely to develop after Live Donor Liver Transplantation (LDLT) than after whole-graft cadaveric OLT3. Therefore, the latter is also associated with increased risk of late HAT.
Although the clinical features of early HAT are well described, the features of delayed HAT are less clearly defined1: the patient may present with manifestations of biliary sepsis or may remain asymptomatic for years. Right upper quadrant pain has been reported to occur in both immediate and delayed HAT. The clinical presentations may include recurrent episodes of cholangitis, cholangitis with a stricture, cholangitis and intrahepatic abscesses, and bile leaks1. Doppler ultrasonography has been extremely sensitive for the detection of HAT in symptomatic patients during the immediate postoperative period but becomes less sensitive as the interval between transplantation and diagnosis of HAT increases because of collateral arterial flow4.
3D gadolinium-enhanced MRA provides excellent visualization of arterial and venous anatomy with a fairly high technical success rate. MRA is a useful adjunct in patients with indeterminate ultrasonography examination in patients who have renal insufficiency or who have allergy to iodinated contrast 5.
Antiplatelet prophylaxis can effectively reduce the incidence of late HAT after liver transplantation, particularly in those patients at risk for this complication6. Vivarelli et al reported an overall incidence of late HAT of 1.67%, with a median time of presentation of 500 days; late HAT was reported in 0.4% of patients who were maintained on antiplatelet prophylaxis compared to 2.2% in those who did not receive prophylaxis6. The option of performing thrombolysis remains controversial. Whether thrombolysis is a definitive therapy or mainly a necessary step in the proper diagnosis of the exact etiology of HAT depends mostly on the particular liver center and needs further analysis7. Definitive endoluminal success cannot be achieved without resolving associated and possible instigating underlying arterial anatomical defects. Reestablishing flow to the graft can unmask underlying lesions as well as assess surrounding vasculature thus providing anatomical information for a more elective, better plan and definitive surgical revision7. Whether surgical revascularization compared to retransplantation is a viable option or only a bridging measure to delay the second transplantation has been a longstanding controversy in the treatment of HAT.
Biliary or vascular reconstruction do not increase graft survival and ongoing severe sepsis at the time of re-graft results in poor survival7. However, although uncommon, delayed HAT is a major indication for re-transplantation7. In the absence of hepatic failure, conservative treatment appears to be effective for patients with hepatic artery thrombosis.
C. perfringensis an anaerobic, gram-positive rod frequently isolated from the biliary tree and gastrointestinal tract. Inoculation of Clostridium spores into necrotic tissue is associated with formation of hepatic abscess8.
Necrotizing infections of the transplanted liver are rare. There have been around 20 cases of gas gangrene or necrotizing infections of the liver reported in the literature. Around 60% of these infections were caused by clostridial species with C. perfringens accounting for most of them. Around 80% of patients infected with Clostridium died, frequently within hours of becoming ill9,10. Those who survived underwent prompt retransplantation and the infection had not resulted in shock or other systemic changes that significantly decreased the likelihood of successful retransplantation8.
Because the liver has contact with the gastrointestinal tract via the portal venous system, intestinal tract bacteria may enter the liver via translocation across the intestinal mucosa into the portal venous system. Clostridial species can also be found in the bile of healthy individuals undergoing cholecystectomy9,10.
The donor liver can also be the source of bacteria. Donors may have conditions that favor the growth of bacteria in bile or the translocation of bacteria into the portal venous blood. These conditions include trauma to the gastrointestinal tract, prolonged intensive care unit admissions, periods of hypotension, use of inotropic agents, and other conditions that increase the risk of potential infection 8,9,10. C. perfringens sepsis in OLT recipients has been uniformly fatal without emergent retransplantation. Survival from C. perfringens sepsis managed without exploratory laparotomy or emergency treatment has been extremely rarely reported8. In those patients who survived, and in whom the infection has not resulted in shock or multiple organ failure, retransplantation may be successful8.
Although our patient survived his intensive care course, his recovery was tenuous as he quickly developed additional hepatic abscesses that led to his eventual demise. Post-mortem examination in our patient revealed intra-hepatic presence of Clostridium perfringens.
He was managed conservatively since he markedly improved both clinically and by liver function tests. Because of this, retransplantation was delayed. He was also already on antiplatelet prophylaxis.
Conclusion:
We report an interesting case of Clostridium perfringens hepatic abscess due to late HAT following OLT. Although the patient initially improved with non-surgical treatment, he eventually died. In similar cases, besides aggressive work-up and medical management retransplantation may be necessary for a better long term outcome.
A 78 year old man was admitted for re-fashioning of a prolapsing colostomy. Nine months previously he had undergone a juxtarenal aortic aneurysm repair complicated by ischaemic colitis, for which he had a sigmoid colectomy and a further laparotomy for refashioning of the stoma.
His past medical history consisted of anaemia, stable angina, superficial bladder cancer, Stage 4 chronic kidney disease, type 2 diabetes mellitus and osteoarthritis. He weighed 77 kg and his exercise tolerance was 100 yards, with the aid of a stick. His medications included doxazosin, quinine sulphate, perindopril, simvastatin, ferrous sulphate, isosorbide mononitrate and aspirin.
On examination, he was apyrexial with a blood pressure of 170/70 mm Hg, a heart rate of 65 bpm, a respiratory rate of 14 bpm and Sa02 of 98% on room air. Serum laboratory tests showed a prothrombin time of 13.4 sec (normal range 9.0 – 13.0 sec), haemoglobin 10.2 g/dL (13.0 – 16.7 g/dl), platelets 131 x109/L (150 – 400 x109/L), sodium 139 mmol/L, potassium 4.2 mmol/L, urea 15.7 mmol/L (2.5 – 7.0 mmol/L), creatinine 196 umol/L (50 – 130 umol/L) and eGFR 29 ml/min/1.73m2 (>60ml/min/1.73m2).
His preoperative pulmonary function tests showed an FVC of 2.78L (93.8% predicted), a reduced FEV1 of 1.66L (75.2% predicted) and FEV/FVC of 59.87%. His ECG showed normal sinus rhythm. A CPEX test taken 12 months previously showed moderately reduced peak aerobic capacity, with a peak V02 of 11 ml/kg/min and an anaerobic threshold (AT) of 7 ml/kg/min.
In the anaesthetic room, the patient was connected to standard monitoring in accordance with AAGBI Guidelines and venous access secured with an 18 g biovalve cannula. Following pre-oxygenation, anaesthesia was induced using fentanyl 200 mcg, midazolam 2 mg, propofol 120 mg and atracurium 50 mg. Tracheal-intubation was achieved (grade 1 view) using an 8.0 mm cuffed endotracheal tube (lo-contour). A TAP block was administered following induction using ultrasound guidance (Sonosite ‘micromax’). A 50 mm insulated Stimi-Plex needle was used to inject a total of 40 ml levobupivacaine 0.375% bilaterally. Anaesthesia was maintained using a mixture of air, oxygen and sevoflurane (1.3 – 2.3 ETAA range). Intravenous (IV) paracetamol 1 g was given intraoperatively. The prolapsing colon was dissected through a circumstomal incision. A 10 cm length of colon was resected after ligation and division of the mesenteric vessels. The new end colostomy was fashioned with interrupted mucocutaneous sutures. Blood loss was minimal and there were no intraoperative complications. The duration of surgery was 1.5 hrs.
The patient spent 30 minutes in the postoperative recovery unit. He was awake, orientated and pain free throughout this period and clinical observations were stable. The patient remained pain-free on the ward and did not require any postoperative opioids. He was medically fit for discharge the following day.
Discussion
There are no previous case reports to our knowledge describing the use of TAP blocks in a patient with such poor CPEX testing preoperatively (AT = 7 ml/kg/min). CPEX testing has been shown to be an independent predictor of morbidity, mortality and length of hospital stay after major abdominal surgery.1 The anaerobic threshold marks the onset of anaerobic metabolism as a result of inadequate oxygen delivery. It is not affected by patient effort and therefore provides a reliable patient specific measurement of functional capacity.2 An AT of at least 11 ml/kg/min is recommended to safely undertake major surgery.2 A combination of an AT of <11 ml/kg/min with ECG evidence of myocardial ischaemia is associated with high mortality and poor outcome. 3One study showed a mortality of 42% in those with ischaemic heart disease (IHD) and an AT <11 ml/kg/min, compared with just 4% in those with no IHD.4
Postoperative morbidity and mortality most often occurs in patients with pre-existing cardiorespiratory disease and a reduced functional capacity, due to their inability to withstand the additional physiological demands placed upon them by major surgery. Many of these patients develop features of organ hypoperfusion due to poor cardiorespiratory reserve.5
Our patient had an AT of 7 ml/kg/min and poor respiratory reserve (exercise tolerance of 100 yards, FEV/FVC 59.87% of predicted) but underwent uneventful intra-abdominal surgery with a good recovery and short length of hospital stay. Contributing factors may have been the anaesthetic technique used, as well as the surgical approach via a parastomal incision. Good intraoperative and postoperative control of cardiovascular parameters, temperature and pain are well known to reduce the surgical stress response and postoperative morbidity, and to improve postoperative outcome.6 Our patient was cardiovascularly stable throughout the perioperative period. His pain was well controlled with no opioid requirements due to the use of bilateral TAP blocks, and this probably contributed to his uneventful recovery, with no critical care requirement.
CPEX testing is not universally accepted as a useful preoperative assessment tool. In studies assessing it as a reliable predictor of outcome, there is heterogeneity in the degree of clinician blinding used. Blinding was used in some studies,1 whilst in other instances, clinicians were aware of the CPEX results and changed their management accordingly. This obscures the true relationship between patient outcome and CPEX-derived measures of risk.7
TAP blocks were first described in 20018 and have been shown to significantly reduce postoperative morphine consumption following abdominal surgery by up to 70%. They reduce pain scores at rest and during mobilisation in the early postoperative period (0-6 hours), and in the first 24 hours.9 The reduced requirement for morphine also leads to a reduction in postoperative nausea, vomiting and sedation.9 It may be possible that the ultrasound guided TAP block confers advantages in procedures with moderate surgical trauma to minimize pain and reduce opioid usage, thereby promoting faster recovery and discharge.8 TAP blocks were the chosen method of analgesia in our patient as they would elicit the least physiological disturbance, but would provide good postoperative analgesia, without opioid-related side effects. This was particularly beneficial, as his pre-existing renal impairment put him at increased risk of opiate toxicity. TAP blocks eliminate somatic pain relating to the surgical incision but do not treat visceral pain. However, our patient tolerated a 10 cm bowel resection with bilateral TAP blocks and intravenous paracetamol. A similar effect has been observed in other studies but the mechanisms behind it are unclear. One theory is that there is an analgesic effect due to high systemic levels of local anaesthetic.10
The use of CPEX testing to determine fitness for surgery should be interpreted with caution as newer anaesthetic and surgical techniques develop. Our patient had IHD and an AT which showed a significantly increased level of risk. However, a combination of regional anaesthesia and a cardio stable general anaesthetic with minimally invasive surgery, allowed a rapid and uneventful recovery with no opioid requirements and a short length of hospital stay.
First described in 1971 by Spillane et al.1, painful legs and moving toes (PLMT) is a syndrome consisting of pain in lower legs with involuntary movements of the toes or feet. Pain varies from moderate discomfort to diffuse and deep and usually precedes movements by days to years. The movements themselves are often irregular and range from flexion/extension, abduction/adduction to clawing/straightening and fanning/ circular movements of the toes.1,2 This syndrome may affect one leg or spread to involve both legs.3
PMLT incidence and prevalence remain largely unknown since it is still a relatively rare disorder worldwide. Age of onset is between the second and seventh decades of life. It has been postulated that lesions of the peripheral or central nervous system after nerve or tissue damage might lead to impulse generation that subsequently causes the symptoms seen in PLMT.4 We report a case of PMLT that presented to our Neurology Movement Disorder Clinic along with a discussion on the pathophysiology, differential diagnosis and clinical management of this rare debilitating condition.
Case report
63 year old, morbidly obese (BMI 41.7) Caucasian male patient with past medical history of stroke 10 years ago, on long term anticoagulation, hypertension, type II controlled diabetes mellitus, asbestos exposure, bilateral hip and knee osteoarthritis, left total knee replacement 2 years ago, and non-traumatic ruptured Achilles tendon; presented with complaints of involuntary movements in both legs over the last 8-10 years. He had unprovoked flexion and extension of the toes along with feet movement at all times with no diurnal variation. He admitted to having a constant severe pain described as 'twisting a rubber band' with 10/10 intensity that radiated up to his calf accompanied by numbness and dorsal swelling of both feet for many months. He claimed to have partial relief whilst walking but had difficulty walking without a cane as he ‘“could not balance with constantly moving [his] feet”’. Tylenol 500mg as required and amitriptyline 20mg at night prescribed by his primary care physician provided no relief.
He also has a history of snoring, daytime fatigue, and non-restorative sleep with frequent nocturnal awakenings due to bilateral feet pain. He recalled having a stroke with transient confusion and focal hand weakness along with visual problems about 10 years previously. All laboratory and radiological investigations were negative and he recovered fully. He had previously served with the US armed forces and had been exposed to ‘Agent Orange’ in Vietnam.
He had no medical allergies and his current medications include amitriptyline 25mg at night, hydrochlorothiazide 25mg once daily, lisinopril 10mg once daily, loradatine 10mg once daily, metoprolol tartrate 20mg twice daily, simvastatin 20mg once daily, vitamin B complex one tablet once daily and warfarin once daily. He denied any history of alcohol, tobacco, or recreational drug abuse in the past. His mother had a history of hypertension and chronic low back pain; no members of his family had any neurological or movement disorders.
Physical examination revealed an alert, awake, and well oriented male with bilateral lower extremity varicose veins. He was observed to have semi-rhythmic flexion-extension and occasionally abduction movements of the phalanges, especially in the great toes. There was a profound decrease in vibration sense below both knees and it was almost absent on both feet, decreased reflexes in both feet, and absent proprioception in the phalangeal joints. He was also observed to have decreased pinprick and monofilament sensation in both legs below the knee. Bilateral ankle reflexes were diminished with negative Babinski sign. Both lower extremity dorsalis pedis and posterior tibial pulsations were palpable. He did not have any cerebellar signs. He did have pitting oedema up to his shins in both lower extremities, extending from his feet to upper one third of the legs. There were no abnormalities noted on the bilateral lower extremity EMG and there was no electrodiagnostic evidence of large-fiber neuropathy.
He was diagnosed with painful legs and moving toes syndrome and started on a trial of gabapentin 300mg at night. He was advised to increase it to 1200mg and to continue taking his amitriptyline 25mg at night. Scheduled MRI of the brain could not be done due to his morbid obesity. He was arranged follow up in three months in the clinic.
Methods
A review of published literature on PLMT was done using MEDLINE and PubMed databases. Searches were conducted to find articles from 1971 – 2010. Medical subject headings used to search the databases included PLMT with subheadings of Movement disorder, Electromyography, and Polysomnography as well as keyword search using ‘PLMT’. Single author reviewed titles and abstracts of potentially relevant articles.
Review of current literature
We reviewed approximately 19 PLMT articles that have been published to date with a total of 72 patients: 30.5% males, 69.5% females (median age 55 & 64 years,
respectively). Clinical presentations in the majority of the cases were burning pain in lower extremities and involuntary movements of the toes. The most common predisposing conditions were neuropathy and radiculopathy (see Table 1).
Table 1 - Painful Legs & Moving Toes Syndrome ~ Review of Literature (1971- 2010)
Author
Year
Sex/ Subjects
Subject age
# of cases
Clinical presentation
Spillane et al
1971
M (4) F (2)
51, 52, 52, 53 66, 68
6
Burning/throbbing LE pain followed by writhing/clawing and flexion/extension movements of the toes
Dressler et al
1994
M (4) F (16)
28, 36, 54, 73 28-76
20
Pain in LE followed by involuntary flexion/extension and abducion/adduction of the toes
Shime et al
1998
F (1)
63
1
Involuntary flexion/extension of the toes bilaterally and aching/crampy pain in both feet
Schott et al
1981
M (1) F (4)
66 56, 57, 69, 77
5
Crushing pain in both feet followed by involuntary writhing and flexion/extension of the toes; burning pain in foot followed by writhing toe movements
Montagna et al
1983
M (1) F (2)
57 74, 76
3
Burning pain in one or both LE followed by involuntary flexion/extension, abduction/adduction, and fanning/clawing of the toes
Shime et al
1998
F (1)
63
1
Involuntary flexion/extension of the toes bilaterally and aching/crampy pain in both feet
Villarejo et al
2004
M (1)
66
1
Paresthesias/burning pain in both feet followed by involuntary flexion/extension and abduction/adduction of the toes
Aizawa et al
2007
F (1)
73
1
Tingling pain in both feet followed by involuntary abduction/adduction of the toes
Guimaraes et al
2007
M (1)
60
1
Wringing-like pain in in L foot and R leg followed by flexion/extension and abduction/adduction of the toes
Eisa et al
2008
M (1) F (1)
62 76
2
Burning pain in bilateral LE followed by semirhythmic flexsion/extension of the toes
Alvarez et al
2008
M (6) F (8)
25-84 (mean 69)
14
Burning pain of LE followed by involuntary flexion/extension, abduction/adduction, fanning, or clawing of the toes
Tan et al
1996
F (1)
57
1
Severe burning pain in both LE followed by involuntary flexion/extension and abduction of the toes
Dressler et al
1994
M (4) F (16)
28, 36, 54, 73 28-76
20
Pain in LE followed by involuntary flexion/extension and abducion/adduction of the toes
Yoon et al
2001
F (1)
56
1
Burning pain in R foot with flexion and lateral deviation of the toes
Miyakawa et al
2010
M (1) F (1)
36 26
2
Burning pain in R arm followed by involuntary flexion/extension of R thumb; pain in L leg accompanied by flexion/extension and abduction/adduction of L toes
Schoenen et al
1984
M (2) F (4)
49, 74 68, 69, 71, 80
6
Burning/aching pain in LE followed by involuntary flexion/extension and writhing of the toes
Sanders et al
1999
F (1)
76
1
Deep/throbbing pain in L leg followed by invloluntary flexion/extension and abduction/adduction of L toes
Ikeda et al
2004
F (1)
75
1
Involuntary flexion/extension of the toes bilaterally followed by pain in both legs
Kwon et al
2008
F (1)
75
1
Painless wriggling movements of the toes in both feet
Total Number of articles reviewed = 19 Total Number of Cases: Male = 22 (Median Age = 55 years); Female = 50 (Median Age = 64 years) Author/Article References in chronological order (Top to below): 1, 2, 3, 4, 5, 6, 7, 8, 9, 11, 16, 17, 23, 24, 25, 29, 31, 32, 33
In 1981 Schott GD et al reported that in 3 PLMT patients the EMG revealed evidence of denervation in the affected muscles. Montagna et al of the University of Bologna, Italy reported 3 cases of PLMT that exhibited evidence of peripheral neuropathy on EMG. Polysomnography (PSG) studies on these patients showed reduced movements during sleep with increase in slow wave or rapid eye movement sleep.5 This suggested the movements could have arisen centrally.
Guimaraes et al of the Universidade Nova Lisboa, Portugal reported one patient with a history of Hashimoto’s disease whose lower extremity EMG showed spontaneous arrhythmic bursts of the affected muscles during wakefulness which disappeared during sleep6. Both suggested the movements could have arisen centrally.
Alvarez et al of the Mayo Clinic described 14 cases of PLMT in 2008 in which burning pain often preceded the movements. PSG studies confirmed these movements would also persist in light stages of sleep which pointed to a central origin.7 Eisa et al of Yale University School of Medicine, Connecticut described 2 cases of PLMT in which one patient had a past history of lumbosacral root injury and the other systemic lupus erythematosus with peripheral neuropathy on EMG.8 Interestingly, in the latter patient her pain occurred years after the onset of involuntary toe movements.8
Discussion
Spinal cord and cauda equina diseases, neuropathies, radiculopathies, drugs and other systemic diseases are the main cause of this syndrome although many cases are still idiopathic. The most common predisposing conditions were neuropathy (i.e. polyneuropathy from alcoholism, hypertrophic mononeuritis, or tarsal tunnel syndrome) and radiculopathy.7 Other etiologies include nerve root lesions, peripheral nerve trauma, spinal ganglia lesions, cauda equina lesion, Wilson’s disease, herpes zoster myelitis, HIV, neuroleptics, and chemotherapeutic agents.9-19
The involuntary movements appeared bilaterally in the toes in our patient, which suggests that central reorganization (especially in the spinal level) is the cause of PLMT. EMG and nerve conduction studies have proven helpful in demonstrating spontaneous arrhythmic bursts of affected muscles and underlying neuropathy in some patients. Although the exact mechanism remains elusive, it has been proposed that impulses generated in lesioned peripheral nerve, posterior nerve root/ganglion, or afferent fibers pass into the spinal cord - some to higher areas to cause pain, while others into the local interneuron and motor neurons to generate involuntary movements of the toes.5
In patients with clinical or electrophysiological evidence of peripheral nerve or root problem, these lesions can initiate or even alter afferent input to the spinal cord and cause subsequent central and efferent motor reorganization, which may explain the limited
success these patients had with nerve blocks or lumbar sympathetic blockade.2 Similarly, some have suggested that even though the radiation of pain following local trauma seemed to resemble causalgia,20 there was a lack of hyperpathia and changes in the soft tissue, bones, and blood vessels as well as a poor response to sympathetic blockade, thus making clinical features of PLMT inconsistent with known radicular disorders.3
Interestingly, some believed that the central nervous system played an essential role in PLMT via a central oscillator.21 It has also been proposed that hyper-excitability of the damaged peripheral nerves could cause symptoms of PLMT by way of the sympathetic nervous system. More specifically, the sympathetic nervous system could potentially serve as a bridge between injured afferent fibers and sympathetic nerve fibers,22 allowing abnormal afferent impulse to travel to efferent fibers and ultimately leading to continuous pain with involuntary movements. This was evident in the fact that lumbar sympathetic ganglion blockade provided moderate symptomatic relief for some patients even though it was short-lived.4 Interestingly, one of the explanations put forth was the possibility of spinal/supraspinal reorganization,23 which coincided with the hypothesis of central reorganization mentioned above.
Clinical Management
Numerous treatments including antiepileptics, benzodiazepines, antispasmodic agents, and antidepressants have been tried with little success.1,2,24,25 However, temporary success was observed with local anesthetic nerve blocks, epidural blocks, sympathectomy/sympathetic blockade, neurectomies, botulinum toxin type A injection, transcutaneous electrical nerve stimulation, vibratory stimulation, and epidural spinal cord stimulation.1,2,15,26,27 Analgesics, steroids, anti-inflammatory agents, vitamin B12 injections, propranolol, quinine sulphate, and local anesthetics only offered temporary relief as well.3 GABAergic agents such as gabapentin and pregabalin were the most effective in attenuating the pain and the movements, possibly via both central and peripheral mechanisms.7,24,25 It has been reported that gabapentin as high as 600mg three times daily could control symptoms of PLMT long-term.25
Treatment of PLMT has also been attempted with botulinum toxin A at the level of lumbosacral roots and peripheral nerves with moderate relief of symptoms, although toe movements did return after a few months.8 It was suggested that botulinum toxin A might have acted via reduction of muscle spindle discharge leading to decreased central sensitization, as well as antisympathetic, antiglutamergic, or anti-inflammatory effects.28
Differential Diagnosis
The syndrome of PLMT exhibits certain features similar to the restless leg syndrome (RLS). In RLS the sensation in the legs could be burning, creeping, or tingling coupled with an urge to move them, especially early in the night. Movements such as walking or stretching relieve the symptoms whereas rest makes them worse. However, in PLMT pain is severe, constant, unrelated to the sleep-wake cycle, and is not relieved by
movements or walking.23 In addition, its involuntary movements of the toes or feet also differ from the myoclonic jerks of RLS.
In conditions such as thalamic syndrome and limb pain with myoclonus, patients may experience pain and involuntary movements as well but they often occur simultaneously as opposed to in PLMT where pain often precedes the movements.17 In disorders such as Parkinson’s disease and dystonia, sustained involuntary movements in the feet can be present and pain can be an associated feature. But the movements are typically sustained muscle contractions, which are different from the typical movements associated with PLMT.
Prognosis
PLMT is a newly discovered syndrome and since there has not been a systematic study following these patients long-term, it is currently quite difficult to predict the outcome of this syndrome and its effect on lifespan, though there has yet been a report of a patient actually dying from this syndrome. However, it is known that PLMT is a debilitating condition that greatly reduces patients’ quality of life.
Conclusion
Since Spillane et al first described it in 1971, there have been more reported cases of PLMT and its variants over the years. Though much progress has been made in elucidating its etiology, its exact mechanism still remains a mystery. Similarly, even though EMG and nerve conduction studies have proven helpful in demonstrating spontaneous arrhythmic bursts of affected muscles and underlying neuropathy in some patients, diagnosis of PLMT remains largely on history and clinical presentation.
Physicians should be aware of this rare debilitating condition. It is important to consider PLMT in a patient with painful legs and/or restless leg syndrome without any significant history of neurological disease or trauma. Treatments such as different combinations of medications and invasive techniques are complex and generally lead to a poor outcome.
Thyrotoxic periodic paralysis (TPP) is an uncommon disorder characterised by simultaneous thyrotoxicosis, hypokalaemia, and paralysis that occurs primarily in males of South Asian descent.1 Many affected patients do not have obvious symptoms and signs of hyperthyroidism and hence may be misdiagnosed or overlooked on presentation.2 We hereby report a male patient who presented to us with weakness of all four limbs. The patient was evaluated and diagnosed to be having TPP.
Case History
A 30-year-old male patient, who was an agriculturist by profession, presented with weakness of all four limbs of one-day duration. The weakness first appeared in his lower limbs and then in the upper limbs. There were no sensory symptoms or bladder involvement. He was not a known hypertensive, diabetic or thyrotoxic patient. He was not on any medication for any significant illness.
On general physical examination, there was no pallor, icterus, cyanosis, clubbing, lymphadenopathy or pedal oedema. Multinodular goitre was noted on thyroid examination. There was no exopthalmos, lid lag, pretibial myxoedema or other signs of thyrotoxicosis. Thyroid bruit was absent. Pulse rate was 96/minute, blood pressure of 140/80mmHg, and respiratory rate 18/minute. On central nervous system examination, the higher mental functions and cranial nerve examination were within normal limits. Motor system examination showed the presence of flaccid quadriparesis with areflexia. Sensory system examination was within normal limits. Cardiovascular and respiratory system examination were normal.
Investigations revealed: haemoglobin (Hb) -13.1 gm%, total count (TC) - 11,400/cmm, platelet count - 49,000/cmm, random blood sugar (RBS) - 110mg/dl, blood urea - 29 mg/dl, serum creatinine - 0.8 mg/dl. Serum electrolyte profile showed sodium - 143 mEq/L, potassium - 2.2mEq/L, chloride - 112mEq/L. Serum calcium and magnesium levels were within normal limits. Electrocardiogram (ECG) was normal. Human Immunodeficiency Virus (HIV) ELISA was non reactive. Bone marrow biopsy and ultrasonography of abdomen were normal. Fine Needle Aspiration Cytology (FNAC) of thyroid showed features of hyperplastic colloid goitre. Ultrasonography of thyroid showed hyperechoic small nodules in both lobes as well as isthmus suggestive of multinodular goitre. Thyroid profile was: total T3 - 2.34 (normal: 0.60 - 1.81ng/ml), total T4 - 13.9 (normal: 4.5 - 10.9 mcg/dl), thyroid-stimulating hormone (TSH) - 0.01 (normal: 0.35 - 5.5IU/ml). Antithyroid antibodies and antiplatelet antibodies were negative. Nerve conduction study was normal. A final diagnosis of TPP with idiopathic thrombocytopenia was made.
The patient was administered 40mmol potassium chloride intravenously. He was treated with tablet carbimazole 10mg three times a day and tablet propanolol 10mg twice a day. The patient’s weakness in all four limbs improved dramatically within an hour after potassium chloride administration. As he had persistent thrombocytopenia during his stay in hospital, he was commenced on tablet prednisolone (1mg/kg body weight). His platelet count normalized in one month after which the steroid dose was tapered and stopped.
Discussion
TPP is an uncommon disorder characterised by simultaneous thyrotoxicosis, hypokalaemia and paralysis that occurs primarily in males of South Asian descent. The overall incidence of TPP in Chinese and Japanese thyrotoxic patients is 1.8% and 1.9% respectively.3, 4 Sporadic cases have been reported in non-Asian populations such as Caucasians, Afro-Americans, American Indians and Hispanics. With population mobility and admixture, TPP is becoming more common in Western countries. Many affected patients are in the age group of 20 - 40 years and do not have obvious symptoms and signs of hyperthyroidism.5 The attack is characterised by recurrent, transient episodes of muscle weakness that range from mild weakness to complete flaccid paralysis. The proximal muscles are affected more severely than distal muscles. Attacks usually first involve the lower limbs, and progress to the girdle muscles and subsequently the upper limbs. Sensory function is not affected. Although patients can present with quadriparesis that resembles Guillain-Barre Syndrome or transverse myelitis, the bladder and bowel functions are never affected. Patient may experience recurrent episodes of weakness that last from a few hours up to 72 hours with complete recovery between the attacks. In the majority of patients, deep tendon jerks are markedly diminished or absent although some patients may have normal jerks.
Patients with TPP usually experience the attacks a few hours after a heavy meal or in the early morning hours upon waking. More than two-thirds present to the emergency department between 2100 and 0900 hours; hence it was initially described as nocturnal palsy or night palsy.6 It has been shown that plasma glucose and insulin responses to meals are markedly higher in the evening than in the morning in control subjects. Such a phenomenon suggests a possible mechanism for the nocturnal preponderance of TPP. Another explanation could be the circadian rhythmicity of many hormones reaching their peak levels during sleep. Hypokalaemia is considered to be the most consistent electrolyte abnormality in TPP and a hallmark of the syndrome along with hyperthyroidism. It has been demonstrated that hypokalaemia is a result of potassium shift into cells and that it is not caused by total body potassium depletion.7 Patients with thyrotoxic periodic paralysis have an underlying predisposition for activation of Na+/K+-ATPase activity either directly by thyroid hormones or indirectly via adrenergic stimulation, insulin or exercise. Increased Na+-K+ ATPase activity is postulated to contribute to hypokalaemia.8
The majority of cases of hyperthyroidism associated with thyrotoxic periodic paralysis are due to Graves disease although other conditions including thyroiditis, toxic multinodular goitre, toxic adenoma, TSH secreting pituitary tumour, ingestion of T4 and inadvertent iodine excess have also been implicated.9 Assaying of thyroid function in patients with hypokalaemic paralysis distinguishes thyrotoxic periodic paralysis from other forms of hypokalaemic periodic paralysis. Thyrotoxic periodic paralysis occurs only in the presence of hyperthyroidism and is abolished when thyroid hormones are normalised.
Immediate therapy with potassium supplementation and beta-adrenergic blockers can prevent serious cardiopulmonary complications and may hasten recovery of periodic paralysis.10 Potassium chloride is given intravenously and/or orally. Regular potassium supplementation as prophylaxis against further paralysis when the patient has normal serum potassium level is ineffective. Effective control of hyperthyroidism is indicated to prevent recurrence of paralysis.
Conclusion
To conclude, although the association of thyrotoxicosis and periodic paralysis has been well known, TPP is often not recognised when first seen because of lack of familiarity with the disorder and partly because of the subtleness of thyrotoxicosis. When a young male of South Asian descent is initially seen with severe lower limb weakness or paralysis, TPP should be considered in the differential diagnosis and investigated for its presence since it is a curable disorder that resolves when euthyroid state is achieved.
An 80-year old lady was referred to a gastroenterology clinic in August 2009 with deranged liver function tests; alkaline phosphatase 180 IU/L (35-120), alanine transferase 147 IU/L (<40), gamma glutamyl transferase 384 IU/L (<45) and globulins 45 g/L (20-35). She had initially presented to her general practitioner with symptoms of lethargy and malaise four months previously. She denied any symptoms of obstructive jaundice and there were no risk factors for hepatitis; she seldom consumed alcohol.
Past medical history included osteoarthritis, migraines and recurrent urinary tract infections; these had been investigated by urology and the patient had undergone cystoscopy and urethral dilatation in September 2003; despite this she continued to experience urinary tract infections and was therefore commenced on prophylactic nitrofurantion by her General Practitioner with approval by the Urologist. This was initially commenced at 50mg at night. This regime was continued for approximately three years however during this time she had a further three treatment courses of nitrofurantoin. In October 2005 her prophylactic dose was therefore increased to 100mg at night. Other medication included lansoprazole 30mg daily, pizotifen 500 micrograms at night, metoprolol 100mg twice daily, simvastatin 10mg at night, senna 15mg at night and furosemide 40mg daily.
On examination there was evidence of palmar erythema and Dupuytren’s contractures but no other stigmata of chronic liver disease. The liver was tender and palpable 4 cm below the costal margin. A liver ultrasound was performed which was normal. Liver screen and autoimmune profile are shown in table 1; notably a positive nuclear antibody was found (1 in 1280 IgG) with Hep 2 cell staining showing a homogenous (ANA) pattern at 1:320 IgG, and a nuclear lamin pattern at 1:1280 IgG;. Due to the positive ANA and raised globulins a suspected diagnosis of nitrofurantoin-induced autoimmune hepatitis was made and a liver biopsy performed.
Liver biopsy (figure 1) indicated a moderate hepatitis which was mainly portal based with multifocal interface hepatitis; these morphological appearances were consistent with those of an autoimmune hepatitis. The patient was advised to immediately stop nitrofurantoin and was commenced on prednisolone 30mg which caused a rapid improvement in LFTs (figure 2). This improvement was maintained following a step-wise reduction in steroid dose and prednisolone was discontinued after eight months of treatment. LFTs are currently normal one month following cessation of steroids
Discussion
This case raises two points of discussion. The first is whether the long term use of nitrofurantoin as prophylaxis for urinary tract infections is appropriate and based on solid evidence. Nitrofurantoin has many side effects and is well documented to cause liver derangement1,2,3. The patient described in this case had been taking nitrofurantoin for seven years and had received a large cumulative dose, on the basis that this was effective prophylaxis. The continuous, long term use of antibiotics as prophylaxis for urinary tract infections is debatable. Madersbacher et al4 recommend the use of prophylactic antibiotics but only after or alongside additional measures including behavioural change, the use of topical oestrogens and the use of alternative therapies; this view is supported by the European Association of Urology5.A Cochrane Review6 in 2004 found that antibiotic use did decrease the number of urinary tract infections compared to placebo but only for the duration of treatment; antibiotics do not alter the natural history of the underlying condition7. There is no clear evidence for duration of treatment and any trials have only been continued for six or twelve months6. It has been noted that all antibiotics had a worse adverse event profile compared to placebo. There was no consensus as to which antibiotic should be used although nitrofurantoin has been associated with a greater withdrawal rate6. One study8 comparing nitrofurantoin and trimethoprim revealed no significant difference in recurrence rates or side effects between the two antibiotics, although this involved a lower dose of nitrofurantoin than was used in this case, and a treatment duration of just 6 months. We would argue that due to the side effect profile of nitrofurantoin and the evidence base available, it is not appropriate to continue it for a duration beyond 6 months.
The second discussion point is whether nitrofurantoin was actually the cause for liver derangement in this case. As documented in a recent review article on the diagnosis of drug-induced liver injury, establishing with any certainty whether liver injury is drug induced can be very difficult3. The key issues are whether there is a temporal relationship between the drug and the onset of liver injury, and whether other causes have been excluded. In this case the patient had negative viral serology and a normal ferritin and caeruloplasmin but her positive autoantibodies raise the possibility of autoimmune hepatitis. Guidelines from the American Association for Liver Diseases9 suggests that the diagnosis of autoimmune hepatitis should be made on the following criteria
laboratory abnormalities (serum AST or ALT, and increased serum total IgG or gamma-globulins)
positive serological markers including ANA,SMA, anti-LKM1 or anti- LC1
histological changes consistent with autoimmune hepatitis i.e. interface hepatitis
This case meets these above criteria for autoimmune hepatitis however the presence of nitrofurantoin does confound the issue. Other case reports10 have reported nitrofurantoin to have caused autoimmune hepatitis based on the relationship between the timing of the drug and the onset of biochemical abnormalities. Bjornsson et al11 performed a comparative study of patients with autoimmune hepatitis and found drugs, particularly nitrofurantoin and minocycline were causally implicated in 9% of cases. When they compared the two groups no significant differences were found in the diagnostic parameters of biochemical, serological and histological abnormalities. In fact the only difference was that no drug-induced cases relapsed on withdrawal of steroids whereas nearly two third of those with non-drug-induced hepatitis relapsed. Bjornsson et al therefore argue in favour of autoimmune immune hepatitis being induced by drugs such as nitrofurantoin; rather than particular drugs simply unmasking sporadic cases based on these management differences.
The patient in this case so far has shown no signs of relapse following steroid withdrawal. We believe that this case does represent one of nitrofurantoin-induced autoimmune hepatitis. In view of the above we would urge readers to consider their use of nitrofurantoin for recurrent urinary-tract infection prophylaxis.
Lactic acidosis is an important cause of metabolic acidosis in hospitalised patients. This usually occurs either due to over production or under utilisation of lactate1 . Most cases of lactic acidosis are due to marked tissue hypoperfusion or hypoxia in systemic shock.
Asymptomatic lactic acidosis has been reported previously during acute severe asthma and attributed to fatiguing respiratory muscles, hypoxaemia and liver ischaemia. It has also been linked to β2 agonist therapy in asthma, although lactic acidosis causing increasing dyspnoea in the asthmatic patient has only been recorded rarely.
Case presentation
We present a case of lactic acidosis in a patient with acute severe asthma who did not have any overt signs of sepsis or tissue hypoperfusion.
Mr IL was a 49 years old male who was known to have moderate asthma. He had multiple previous admissions to hospital with exacerbation of asthma but had never required an intensive care admission and had never been intubated. His other comorbidities included atrial fibrillation, ischaemic heart disease and depression.
His usual medications included salbutamol, budesonide and salmeterol inhalers, aspirin, atorvastatin and digoxin. He was a mechanic by trade with no obvious occupational sensitisation. He had no pets at home. He was a smoker with a 20 pack year history. Recent lung function tests showed an FEV1/FVC of 0.68 with a post bronchodilator FEV1 of 4.17 L (95% predicted).
He was admitted with a 1 week history of worsening shortness of breath, dry cough and wheeze. His baseline blood tests including full blood count, C reactive protein, liver and renal function were normal. Chest radiograph was unremarkable. Arterial blood gas showed no evidence of hypoxia or acidosis.He was treated as acute severe asthma with back to back nebulisers, intravenous hydrocortisone and magnesium sulphate resulting in gradual improvement in bronchospasm and peak expiratory flow rate.
Despite optimal treatment, his breathing started to deteriorate. Arterial blood gas at this time showed lactic acidosis with normal oxygenation (Table 1). There was no clinical or biochemical evidence of haemodynamic compromise or sepsis. A presumptive diagnosis of lactic acidosis secondary to salbutamol was made. The nebulisers were withheld and he has transferred to high dependency unit for closer monitoring. The acidosis completely resolved in the following 12 hours on stopping salbutamol and the patient made an uneventful recovery.
Table 1: Serial Arterial Blood Gases (On admission, 4 hours later and on stopping salbutamol)
00:22
04:06
07:42 *
10:50
11:35
12:24
14:29
17:33
23:32
FiO2
100%
60%
60%
60%
60%
40%
40%
35%
28%
pH (7.35-7.45)
7.36
7.28
7.26
7.32
7.34
7.37
7.37
7.39
7.41
pCO2 (4.5-6.0 kPa)
4.87
4.74
4.15
3.31
3.98
3.9
4.7
5.08
5.49
pO2 (11-14 kPa)
27
19.2
16.5
19
18
14.1
12.5
13
11.8
HCO3 (22-28 mmol/L)
22
16.3
13.6
12.4
15.6
16.6
19.9
22
25.6
BE (2- -2)
-2
-9.1
-12
-12
-9
-7.6
-4.4
-1.5
1.4
Lactate (0.5-2 mEq/L)
1.8
7.6
9.7
9.3
7.6
6.8
3.6
1.4
1.1
* Salbutamol witheld
Discussion
Lactate is a product of anaerobic glucose metabolism and is generated from pyruvate. Normal plasma lactate concentration is 0.5-2 meq/L. Most cases of lactic acidosis are due to marked tissue hypoperfusion or hypoxia in systemic shock2 .
Lactic acidosis can occur in acute severe asthma due to inadequate oxygen delivery to the respiratory muscles to meet an elevated oxygen demand3 or due to fatiguing respiratory muscles4 . A less recognised cause of lactic acidosis is treatment with salbutamol. The mechanism of this complication is poorly understood.
Salbutamol is the most commonly used short acting βagonist. Stimulation of β adrenergic receptors leads to a variety of metabolic effects including increase in glycogenolysis, gluconeogenesis and lipolysis5 thus contributing to lactic acidosis.
Table 2 shows an assortment of previously published case reports and case series of lactic acidosis in the context of acute asthma.
Table 2: Details of etiology and consequences of lactic acidosis in previously published case reports
Reference
n
Suggested etiology of lactic acidosis
Effect of lactic acidosis
Roncoroni et al, 1976 [6]
25
Uncertain: increased respiratory muscle production, decreased muscle or liver metabolism
None observed
Appel et al, 1983 [7]
12
Increased respiratory muscle production, decreased muscle or liver metabolism
8 out of 12 developed respiratory acidosis, 6 required invasive ventilation
Braden et al, 1985 [8]
1
β2 agonist, steroid and theophylline therapy
None
O’Connell & Iber, 1990 [9]
3
Uncertain: intravenous β2 agonist versus severe asthma
None
Mountain et al, 1990 [10]
27
Hypoxia and increased respiratory muscle production
None
Maury et al, 1997 [11]
1
β2 agonist therapy
Inappropriate intensification of β2 agonist therapy
Prakash and Mehta, 2001 [2]
2
β2 agonist therapy
Contributed to hypercapneic respiratory failure
Manthous, 2001 [12]
3
β2 agonist therapy
None
Stratakos et al, 2002 [3]
5
β2 agonist therapy
None
Creagh-Brown and Ball, 2008 [13]
1
β2 agonist therapy
Patient required invasive ventilation
Veenith and Pearce, 2008 [14]
1
β2 agonist therapy
None
Saxena and Marais, 2010 [15]
1
β2 agonist therapy
None
Conclusion
In this case, the patient developed lactic acidosis secondary to treatment with salbutamol nebulisers. The acidosis resolved spontaneously without any specific treatment.
Lactic acidosis secondary to β agonist administration may be a common scenario which can be easily misinterpreted and confuse the clinical picture. Acidosis itself results in hyperventilation which could be mistaken for failure to treat the response. This may in turn lead to inappropriate intensification of treatment.
Tumefactive multiple sclerosis (MS) is a rare variant of MS. This form of MS can masquerade as neoplasm or infectious etiology. Understanding of the disease is limited to case report but it is associated with high morbidity and mortality.
Case report
A 44 year old man presented with a 2-month history of progressive right upper extremity weakness, confusion and visual change. Physical exam revealed weakness, hyperreflexia on the right side and right homonymous hemianopia. MRI of the brain showed multiple ring-enhancing lesions located in both cerebral hemispheres. CSF analysis disclosed elevated protein with positive oligoclonal bands and myelin basic protein. Stains and cultures for bacteria and mycobacteria were negative. Serologies including HIV, Toxoplasmosis, and Lyme were all negative. Patient was treated with high-dose IV corticosteroid and clinically improved. One month later, he presented with increasing confusion, aphasia and progressive weakness. Repeat MRI of the brain revealed worsening multiple ring-enhancing lesions with surrounding vasogenic edema in most lesions. High-dose corticosteroid was promptly started. There was also concern about infection, especially brain abscess; hence, intravenous ceftriaxone, vancomycin, and metronidazole were empirically given. Due to uncertainty of diagnosis, first brain biopsy at right frontal lobe lesion yielded non-specific gliosis. Repeat MRI brain showed increasing number of ring-enhancing lesions in both cerebral hemispheres. As a result, a second brain biopsy was performed, which showed an active demyelinating process consistent with multiple sclerosis. Patient experienced severe disability and was discharged to long-term facility with slowly tapered schedule of corticosteroid. He was readmitted several times and eventually family decided hospice care.
Discussion
Multiple sclerosis is diagnosed by demonstrating clinical and/or radiographic evidence of dissemination of disease in time and space1. Tumefactive MS is a term used when the clinical presentation and/or MRI findings are indistinguishable from a brain tumor2. Not all case of tumefactive MS are fulminant. Marburg variant MS is an acute rare variant of MS which has a rapidly progressive course with frequent, severe relapses leading to death or severe disability within weeks to months3. The tumefactive demyelinating lesions are defined as large (>2 cm.) white matter lesions with little mass-like effect or vasogenic edema, and post-gadolinium magnetic resonance imaging (MRI) typically showing an incomplete ring enhancement2,4. The clinical and imaging characteristics of these demyelinating lesions may mimic primary and secondary brain tumors, brain abscess, tuberculoma, and other inflammatory disorders e.g. sarcoidosis, primary sjogren’s syndrome5. As a result, tumefactive MS is frequently misdiagnosed. There are some MRI characteristics that are more suggestive of tumefactive demyelinating lesions than of other etiologies. These include incomplete ring enhancement, mixed T2-weighted iso-and hyperintensity of enhanced regions, absence of a mass effect and absence of cortical involvement2,6. Differential diagnosis of rapidly progressive neurological deficit with ring-enhancing lesions include brain abscess, primary brain neoplasm or brain metastasis, acute disseminated encephalomyelitis (ADEM) and tumefactive multiple sclerosis. Careful clinical history, CSF study, serial MRI evaluation and follow-up are usually sufficient to make a diagnosis. Some cases pose considerable diagnostic difficulty owing to clinical and radiographical resemblance to brain tumor, for which biopsy may be warranted. Pathologically, the lesions are characterized by massive macrophage infiltration, acute axonal injury, and necrosis. No specific histological features distinguished specimens derived from patients developing classic multiple sclerosis from those who had tumefactive form7. A limited number of cases of Marburg’s variant MS have been reported in the literature whereby most patients died within a period of weeks to months. Only two cases survived after one year7,8. There is no current standard treatment for this condition. Plasma exchange and Mitoxantrone are reportedly showed some promising options9,10.
Figure A: FLAIR imaging at first presentation showed lesion in both hemisphere. Figure B: FLAIR imaging at one month later showed progression of multiple lesion in both hemisphere. Figure C: T1 Post contrast imaging showed intense ring enhancement pattern in almost all lesions with mild edema and minimal mass effect. Figure D: Showed lesion view as sagittal section.
Our patient presented somewhat like a stroke with visual field defect and right hemiparesis which is unusual in MS, but MRI and CSF exam yielded a diagnosis of probable MS. Because of his abrupt clinical deterioration and impressive worsening of his MRI, concern was raised about possibility of infection or neoplasm. Hence, he received two brain biopsies, the second of which showed active demyelination, confirming the diagnosis of severe tumefactive multiple sclerosis and can be consider as a Marburg variant multiple sclerosis.
Conclusion
Marburg variant multiple sclerosis carries a high morbidity and mortality. This disease notoriously mimics other conditions leading to delay diagnosis and treatment. Absence of definitive diagnosis test apart from brain biopsy makes diagnosis, prognosis and treatment decisions difficult.
Hypertension is common but, with early detection and treatment, it is rare to see malignant hypertension. We report a patient who presented with signs suggestive of thrombotic thrombocytopenic purpura and severe hypertension, which resolved with the treatment of hypertension.
CASE REPORT:
A 34 year old African American male presented to the emergency department (ED) having experienced nausea, vomiting and diarrhoea for two days. He denied haematochezia, meleana or sick contacts at home. He complained of blurred vision without photophobia, headache and mild chest discomfort. His past medical history was unremarkable. The patient did not have any significant family history. Smoking history was significant for a pack of cigarettes daily for seven years. He reported occasional alcohol intake, and denied use of recreational drugs. On presentation, this patient’s blood pressure was 201/151 mmHg, with a mean of 168 mmHg. Pulse 103 beats per minute, respirations 20 per minute and temperature 98.4F. Physical examination was otherwise unremarkable, including absence of focal neurological deficits.
Blood tests showed: Haemoglobin 12.6 g/dl, White cell count 13.9 g/dl, Platelets 67000, Sodium 136, Potassium 3.4, BUN 24, Creatinine 2.56 and LDH 556. Chest x-ray showed cardiomegaly. A non-contrast computed tomography scan of the brain did not show any sign of stroke (haemorrhage). Urinalysis was positive for proteins 4+, a large amount of blood, 0-2 white blood cells/high power field (HPF) and 0-2 red blood cells/HPF.
Figure 1
The patient’s initial treatment whilst in the ER consisted of a Labetalol drip. His mean arterial pressure decreased to approximately 115 mmHg during the first hour, and his chest pain and headache improved with the control of elevated mean arterial pressure. Furthermore, over the next 24 - 48 hours, the patient’s blood pressure was brought down to 138/86 mmHg and his blurred vision improved significantly. Subsequently, intravenous medications were switched to an oral regimen. Blood peripheral smear from the day of admission was significant for the schistocytes (Figure 1) suggesting ongoing haemolysis. Renal ultrasound was unremarkable. His cardiac ultrasound revealed an enlarged left ventricle, however no valvular abnormality was seen. Serum calcium and thyroid stimulating hormone levels were normal, as were urine catecholamines and vanillylmandelic acid level. On two week follow up in the outpatient clinic, the patient’s platelet count and creatinine had returned back to baseline and peripheral smear did not reveal any schistocytes as the blood pressure came under better control. [Table 1]
Variable
On day 1
Day 3
Day 5
Day 6
Follow-up in 2 weeks
Haemoglobin
12.6
9.3
9.3
10.3
11.4
Platelets
67, 000
65,000
90,000
125,000
204,000
Retic. count
3.9
--
--
4.3
--
Creatinine
3.06
2.86
2.69
2.4
2.3
BUN
29
27
28
27
27
LDH
556
370
333
240
--
Troponin
0.10
0.08
0.06
0.05
--
Peripheral Smear
Schistocytes
--
--
--
No Schistocytes
Table 1
DISCUSSION:
Malignant hypertension is a medical emergency with an incidence of 1% in hypertensive patients1and is more common in the African American population2. Depending on the clinical presentation, it must be differentiated from thrombotic thrombocytopenic purpura (TTP), disseminated intravascular coagulation (DIC), glomerulonephritis and vasculitis.
Suspicion for TTP was initially high in this patient because of haemolysis, thrombocytopaenia, central nervous system (CNS) manifestations and renal insufficiency. However, TTP did not explain the presence of elevated blood pressure3,4nor the improvement in symptoms and signs with the management of this, which clearly supports our diagnosis. Rapidly progressive glomerulonephritis did not explain the CNS symptoms, and a normal prothrombin time and activated partial thromboplastin time ruled against disseminated intravascular coagulation5. The patient did not have a history of preceding diarrhoea6, which could possibly direct towards haemolytic uraemic syndrome (HUS)4. There was no history of prosthetic valves, nor clinical evidence of vasculitis. The patient’s symptoms of severe hypertension, haemolysis, thrombocytopaenia and renal failure were consistent with malignant hypertension, and treating the hypertension7gradually resolved the thrombocytopaenia, haemolysis and renal failure8.
CONCLUSION:
This case report highlights that malignant hypertension is a medical emergency which can present with features resembling a wide variety of diseases, including TTP and HUS. Using appropriate management to control the elevation in blood pressure can help reveal the underlying diagnosis.
Infection of a prosthetic total knee joint is a serious complication1 and should be diagnosed promptly2 and treated aggressively. We present an interesting case of MRSA infection of a primary total knee replacement following an IV cannula infection leading to bacteremia and subsequent infection of the knee prosthesis, complicated by stevens- Johnson syndrome .
There were many challenging issue which are outlined including diagnosis and management.
Case Report
A 63-year- old lady had an elective total knee arthroplasty for severe osteoarthritis of the knee. She had a background history of well-controlled type 2 diabetes mellitus and was on warfarin for a previous pulmanory embolism. As per the hospital protocol her warfarin was stopped before surgery until her INR was <1.5 and she was heparinised with a view of
warfarinizing after the surgery. She had an uneventful knee arthoplasty, but unfortunately one of her IV cannula site became cellulitic. She was empirically started on oral flucloxacillin after taking blood cultures and sending the cannula tip for microscopic culture and sensitivity (which is routinely done has hospital protocol for infected cannula sites).
Surprisingly the tip grew MRSA and also had MRSA bacteraemia. She became systemically unwell and septic, and was treated aggressively with parentral vancomycin for MRSA bacteraemia. She had a transeosphageal echocardiogram to rule out cardiac vegetation. She gradually improved but developed typical papular rashes over her palm, dorsum of hand, extensor surface of arm and forearm and trunk and buccal mucosa (Fig 1 and 2) .
Fig 1: Rash over the dorsum hands
Fig 2: Rash over the extensor aspects of forearm
She had a severe allergic reaction to vancomycin and the skin biopsy of the lesion confirmed that she had developed Stevens-Johnson syndrome. An alternative antibiotic was started following discussion with the specialist bone infection unit. She gradually improved over the next few weeks without any problem in her prosthetic replaced knee. At about 6 weeks post- operatively she developed severe pain and hot swelling of her replaced knee with decrease range of motion. Her inflammatory markers were markedly raised and the knee aspirate confirmed MRSA infection of the total knee replacement. She was referred to a specialist bone infection unit due to the complexity of the case, where she successfully underwent two- stage revision.
Discussion
Infection of a Knee replacement is a serious complication that requires significant hospital-based recourse for successful management3. The rate of infection of a primary knee replacement varies from 0.5- 12%1. Rheumatoid arthritis , previous surgery , diabetes mellitus are all associated with an increased risk of infection 4. Although there is no absolute diagnostic test for peri-prosthetic infection2 , a high index of clinical suspicion is essential. There has been a case report on MRSA cervical epidural abscess following IV cannulation 5, but to the best of our knowledge there has been no previous report of MRSA- infected knee arthroplasty following complications of IV cannulation. Stevens-Johnson syndrome involves rare but severe cutaneous adverse reactions related to a variety of medications including antibiotics6. Parenteral vancomycin is the first line treatment for MRSA bacteraemia. It is recognised that vancomycin is indicated in inducing Stevens -Johnson syndrome, mortalitiy being 30-100%7. It is vital that Stevens- Johnson syndrome is recognised early so that offending agents are stopped and supportive treatment commenced. Early dermatological consultation, skin biopsy and direct immunofluorescence7 are essential to confirm diagnosis so that effective treatment can be instituted.The diagnosis and management of this serious complication is complex and requires considerable recourse allocation by the patient, the hospital, the infectious disease specialist, and the orthopaedic surgeon1,5.
A 73 year old lady presented for assessment of her recurrent right sided pleural effusion. She had a history of gallstones and underwent open cholecystectomy. One month after surgery the patient had recurrent pleural effusion requiring thoracocentesis on a monthly basis. On the chest x-ray, the pleural effusion was seen exclusively on the right side occupying the whole right hemithorax.
The pleural fluid was transudative on multiple occasions and there was no evidence of malignant cells. Her echocardiography revealed preserved cardiac function. An abdominal ultrasound showed findings of cirrhosis and splenomegaly consistent with portal hypertension.
Image 1
Computerised tomography (CT) of the chest and abdomen revealed a large right-sided pleural effusion and minimal ascites (Image 1). An ultrasound guided paracentesis was performed with difficulty and only 17cc of fluid.was obtained. The abdominal fluid showed similar consistency as the pleural fluid. The blood workup at the same time was unremarkable.
Image 2
Intra-peritoneal administration of 99mTc-sulphur colloid was attempted but failed in the absence of ascites. Computed tomography with three dimensional reconstruction at the diaphragmatic level revealed a defect in the posterior aspect of the right hemidiaphragm (Image 2 black arrow) and also revealed irregular contours of the liver, an indirect sign of diaphragmatic defect (Image 2 white arrow).
The patient declined any surgical intervention at that point including the option of pleurodesis. She was started on diuretics and a low salt diet with significant improvement.
Discussion:
Pleural effusion due to hepatic cirrhosis and ascites is well known, but hepatic hydrothorax in the absence of ascites is a rare complication. We report a case of liver cirrhosis with a large and recurring right sided pleural effusion that had an apparent abdominal source in the absence of ascites. We review the characteristics and treatment for hepatic hydrothorax in the absence of ascites.
Hepatic hydrothorax is defined as the presence of significant pleural effusion in a cirrhotic patient without primary pulmonary or cardiac disease1. Postulated mechanisms for the development of pleural effusions in patients with hepatic cirrhosis include: hypoalbuminemia and decreased oncotic pressure leakage of the plasma from the hypertensive azygos vein, lymphatic leak from the thoracic duct, passage of ascitic fluid to the pleural space by way of lymphatic channels in the diaphragm, and transfer of peritoneal fluid directly via diaphragmatic defects2.
The usual unilaterality of hepatic hydrothorax could be attributed to a congenital factor, but not to physiologic mechanisms3. The most likely explanation appears to be that ascitic fluid passes through congenital or acquired fenestrations in the diaphragm directly into the pleural space2. The description of hepatic hydrothorax in the absence of ascites is very rare1. The flow of the ascitic fluid into the pleural space equaled the rate of ascites production in patients with this entity3.
The composition of pleural fluid from hepatic hydrothorax is similar to that of ascitic fluid. Pleural effusions associated with portal hypertension are always transudative1. Nuclear scans can be performed to establish the diagnosis of hepatic hydrothorax with fairly high accuracy. Intra-peritoneal administration of 99mTc-human serum albumin or 99mTc-sulphur colloid can be used to demonstrate the communication between the peritoneal and pleural space. Recent advances in radiological imaging have enabled investigators to examine in detail the diaphragmatic defects responsible for the development of hepatic hydrothorax1.
The management is challenging and frequently associated with poor outcomes in most cases. Dietary restriction of sodium intake and the addition of diuretics is the initial approach. Thoracocentesis can be performed in patients with dyspnoea due to hepatic hydrothorax for immediate relief of symptoms. When thoracocentesis is required too frequently in patients on maximal sodium restriction and optimal diuretics, alternative treatment options must be considered1, 3.
Over the last few years, new insights into the pathogenesis of this entity have lead to improved treatment modalities such as portosystemic shunts (TIPS) and video-assisted thoracoscopy (VATS) for closure of diaphragmatic defects. Both, though temporary measures, are perhaps the best available bridging to liver transplantation in selected patients with refractory hepatic hydrothorax2, 3.
A 52 year-old Caucasian male with a history of chronic obstructive airway disease (COPD) presented to the emergency department complaining of progressive shortness of breath. Two days prior, the patient had presented to the ED with similar complaints that resolved with aerosol treatments and the patient was discharged on a metered dose inhaler (MDI). The patient had been prescribed MDI’s (metered dose inhalers) previously for management of his COPD, but due to financial constraints he had been unable to fill his prescription for the past month. Emergency medical services (EMS) suspected COPD exacerbation and administered 40 mg prednisone IV and two albuterol-ipratropriumnebulisertreatments en route to the hospital, which improved the patient’s breathing symptoms.
Upon arrival to the hospital, his blood pressure was 129/90, respirations 28, pulse 127, and he had an oxygen saturation of 100% on 7L/min. Physical examination revealed increased work of breathing, and wheezes in all lung fields with prolonged expiratory phase. The cardiovascular exam was normal except for tachycardia. A Routine electrocardiogram (ECG) revealed sinus tachycardia and T wave inversions in anterior leads. Chest x-ray showed old scarring in the left lower lobe. Routine cardiac enzymes showed mild elevation with a serum troponin level of 0.68ng/ml (normal range 0.0ng/ml-0.05ng/ml). The second set of troponin peaked at 1.66 ng/ml (normal 0.0ng/ml-0.05ng/ml). In view of the elevated cardiac enzymes atransthoracicechocardiogram was performed which demonstrated multiple wall motion abnormalities and reduced left ventricular ejection fraction of 25%. Coronary angiography demonstrated normal coronary arteries. Left ventriculography revealed hypokinetc mid-anterior and apical segment with a hypercontractile base with reduced ejection fraction (EF) of around 25% (normal range EF 55-65%) (Figure 1)
Figure 1. Left ventriculography demonstrating the classic appearance of Takotsubo cardiomyopathy
In light of the systolic dysfunction not in proportion with the degree of coronary artery stenosis and the multiple areas of wall motion abnormalities seen on echocardiogram, the diagnosis of Takotsubo cardiomyopathy (TCMP) was made. The diagnosis was further supported by the presence of ECG changes, troponin elevation, and the added social stresses of being unemployed. Over the course of his stay in hospital, the patient’s breathing improved with oral prednisone, inhaled tiotropium, and fluticasone/salmeterol. The patient was also treated with an angiotension converting enzyme inhibitor (ACE inhibitor), aspirin, statin, and beta-blockers. There were no adverse coronary events during the course of his hospital stay and the patient was discharged after four days. A Follow up echocardiogram after 4 weeks showed normal left ventricular systolic function.
DISCUSSION
Takotsubo cardiomyopathy (TCMP), also called stress-induced cardiomyopathy, apical ballooning syndrome, or broken heart syndrome, is a transient systolic dysfunction of the ventricles in the absence of significant coronary artery disease. Once thought to be a rare syndrome, TCMP is increasingly being identified in clinical practice, however, the prevalence and incidence are not known. It is estimated that 0.7-2.5% of patients who present with acute coronary syndrome are found to have TCMP1 .The majority of these patients are postmenopausal females, with a mean age of 62-75 years. They may present with chest pain and have a recent history of an emotional stress or severe medical illness. 1
The clinical manifestations of TCMP can mimic those of an acute myocardial infarction. Although, chest pain is a common presenting symptom, patients may also have complaints ofdyspnoeaand arrhythmias. In our casedyspnoeawas the predominant symptom and was easily confused with COPD exacerbation. Recently a few cases of concomitant stress cardiomyopathy with obstructive airway disease have been documented in literature. 2-4 While the pathophysiology of the coexistence of these two disorders is not fully understood, it is thought that both stress induced cardiac dysfunction due to exaggerated sympathetic activation and use of sympathomimetic bronchodilators instigates the myocardial stunning in such patients. Furthermore, an emotional stressor, such as death of a family member, or a physiological stressor, such as an acute medical illness, is thought to be a trigger for cardiomyopathy. 5 It is believed that the syndrome is not a result of anischemia, but there is some evidence to suggest thatoestrogenlevels may have a role in modulating the sympatho-adrenal outflow in TCMP. In mice models, chronic oestrogen supplementation seemed to have protective effects from exaggerated sympathetic outflow from the heart and brain6 . Postmenopausal women with low levels ofoestrogenmay be more vulnerable to the exaggerated catecholamine release in responses to stressors. 7
The characteristic finding in TCMP is a transient mid-ventricular or apical ballooning due to a hypokinetic portion seen on echocardiogram or on a left ventriculography. Systolic dysfunction is usually transient, inconsistent to the perfusion area of a single coronary artery, and usually resolves within 4-6 weeks. 8 Additional findings include ECG changes with ST segment deviations in precordial leads being the most common. Cardiac enzymes have been noted to have moderate elevations.9.
As data regarding the treatment of TCMP is limited, medical management mainly consists of symptomatic therapy with aspirin, ACE inhibitors, beta-blockers, and diuretics, also used in acute coronary syndrome.10 Patients who present acutely are treated as acute coronary syndrome and often receive emergency coronary angiography. However, less invasive imaging techniques, such as echocardiograms, should first be examined carefully. Due to the transient nature of the syndrome, the duration of treatment is unknown with some studies suggesting that there is no benefit with chronic treatment. 11 The prognosis is fairly good, with in hospital mortality rates being reported to range from 0-8%, and recovery of left ventricular function in the majority of patients. 9, 12
TCMP is difficult to distinguish from acute coronary syndrome on first presentation. Our patient had significant social stress. She presented with severedyspnoeaand was treated for COPD exacerbation. Elevation of cardiac enzymes and ECG changes lead to further evaluation and diagnosis of stress cardiomyopathy. This atypical presentation of TCMP showcases the importance ofutilisingthe routine noninvasive imaging and laboratory values to guide the diagnosis. Furthermore physicians need to maintain a high clinical suspicion for this syndrome.
It is often forgotten that smoking inducescytochrome P450 (CYP) 1A2, resulting in altered concentrations and required doses of drugs metabolized by this route. Conversely, upon cessation of smoking, concentrations of these drugs can rise to toxic levels unless appropriate dose adjustments are made. Medical staff, and others involved with smoking cessation counselling, need to be aware of this if potential harm to patients is to be avoided. Here, we describe a patient who developed tonic clonic seizures due to Theophylline toxicity, having ceased smoking 2 weeks earlier.
Case Report
Our patient is a 76-year-old lady who presented to A&E feeling generally unwell, with a two day history of dizziness. Her medical history included atrial fibrillation, which was treated with Digoxin 62.5mcg, and Chronic Obstructive Pulmonary Disease, for which she took a combination inhaler of 25mcg Salmeterol Xinafoate and 250mcg Fluticasone Propionate, 2 puffs twice a day, plus Theophylline MR 175mg twice daily. She had successfully given up smoking 2 weeks prior to admission, having smoked 100 cigarettes per day over the previous 40 years. On admission, she was in atrial fibrillation, well controlled with a heart rate of 90 beats per minute, and normotensive, with no evidence of postural hypotension. Respiratory system examination revealed prolonged expiration, in keeping with COPD, but the rest of the examination was unremarkable. Routine blood investigations, including full blood count, urea, creatinine and electrolytes, liver function tests and C-reactive protein, were all normal apart from a serum potassium level of 3.0mmol/l (NR 3.5-5.0mmol/l). Theophylline concentrations were not tested at this point. A chest X-ray showed hyper-inflated lung fields in keeping with Chronic Obstructive Pulmonary Disease.
The patient was admitted for observation, and treated with Trimethoprim for a presumed urinary tract infection on the basis of urinalysis, which was positive for leukocytes and nitrites.
Two days following admission, the patient developed facial spasms and twitching of her muscles of her upper limbs. Over the next 24 hours, the patient had two witnessed tonic-clonic seizures, which were terminated with intravenous Lorazepam. An urgent CT (Computed Tomography) scan of the brain was performed. This showed changes in keeping with small vessel disease only, nothing to account for new onset of seizures. Following a post-ictal period, the patient recovered, but then the following day had a further two tonic-clonic seizures. It was at this point that a blood Theophylline concentration was requested.
The Theophylline concentration was reported as 41.6mcg/ml (NR 10-19.9mcg/ml), more than twice the upper safe therapeutic concentration. A search of the laboratory system revealed that this patient’s Theophylline concentration had been within the therapeutic range when last checked 2 months prior to admission, while she was still smoking.
The Theophylline was immediately stopped and the patient closely monitored at the Medical High Dependency Unit for further fits, arrhythmias or electrolyte disturbances. Other causes of seizures had been investigated and excluded.
The patient’s neurological symptoms improved dramatically following cessation of Theophylline, with no further twitching, muscle spasms or seizures. Within three days her Theophylline concentration returned to a safe level, but the drug was not recommenced. Unfortunately, however, the patient died from sepsis two weeks following her admission without having left hospital.
Discussion
Theophylline has largely fallen out of favour as a treatment for airways disease due to its very narrow therapeutic index. Even within the therapeutic range, side effects frequently occur. These side effects range from mild, including tremor and gastrointestinal disturbance, to serious and potentially life threatening, such as seizures and cardiac arrhythmias. Following acute overdose, hypokalemia, hyperglycemia, hypercalcemia, hypophosphatemia, and acidosis commonly occur.
Theophylline is mainly metabolised in the liver by demethylation or oxidation using the cytochrome P450 system. The 8-hydroxylation of Theophylline to 1,3-dimethyluric acid (1,3-DMU) via cytochrome P450 1A2 is the major pathway.
The cytochrome P450 enzyme CYP1A2 mediates the rate-limiting step in the metabolism of Theophylline1, andthe polycyclic aromatic hydrocarbons found in cigarette smoke are potent inducers of this enzyme2. For this reason, smokers may need up to double the dose of Theophylline to achieve therapeutic effect compared with non-smokers3.
The relationship between smoking cessation and Theophylline has also been the subject of many studies. Lee et al demonstrated that stopping smoking for 1 week resulted in a 37.6% decrease in clearanceand a 35.8% increase in the half-life of Theophylline, and that the dose needs to be reduced by one fourth to one third after brief abstinence from tobacco to prevent potentially toxic concentrations4. For this reason, careful monitoring of plasma Theophylline concentration should be considered essentialfor optimal dosing in patients following smoking cessation. The study by Faber et al recommends that the dose of CYP1A2 substrates such as Theophylline should automatically be reduced by 10% on cessation of heavy smoking and thereafter be guided by plasma concentration monitoring5.
We found one report in which Theophylline toxicity resulted in a patient’s death following a similar presentation to the one described above6. This highlights the close attention that must be paid to drug concentration monitoring by physicians when advising patients to quit smoking.
Theophylline is not the only drug which needs to be considered when patients stop smoking. Other examples are Clozapine, Olanzapine, Haloperidol and Flecanide to name a few. These drugs are also substrates for CYP1A27.
Conclusion
When advising patients to stop smoking, it is essential that physicians routinely review the drugs that the patient is taking to look for those that may require dose adjustments. In the case of Theophylline, careful monitoring of Theophylline concentrations, for instance weekly in the first few weeks following smoking cessation, is essential to avoid potentially life-threatening complications.
A 44 year old male presented to the emergency department complaining of shortness of breath. The symptoms had commenced suddenly four weeks ago. He had been breathless at rest, and subsequently developed a productive cough with white sputum. He denied chest pain. He was known to have the sickle cell trait but was otherwise in good health. He was a non-smoker.
Since the onset of symptoms, and prior to this admission, the patient presented to two different emergency departments. The working diagnosis was, and remained, community acquired pneumonia. On initial presentation empirical treatment for a community acquired pneumonia was commenced. Failure to improve resulted in additional cover for atypical organisms and the prescription of a short course of steroids on the subsequent admissions.
Initial observations revealed the patient was tachypnoeic and tachycardic, with a respiratory rate of 25 breaths per minute, heart rate of 114 beats per minute. He was apyrexial (temperature of 36.5°C ). Pulse oximetry showed an oxygen saturation of 94% on room air. His blood pressure was recorded as 183/99 millimetres of mercury.
On examination large volume peripheral pulses, raised jugular venous pressure (5 cm), bi-basal crepitations, and bilateral ankle oedema were elicited/identified. Auscultation of the heart revealed a loud diastolic murmur audible throughout the praecordium.
A 12 lead ECG showed normal sinus rhythm, normal axis and left ventricular hypertrophy. Arterial blood gas analysis on room air showed a pH of 7.46, pa02 9.6 kPa, pCO2 4.3 kPa, HCO3 23.8 mmol/L, BE + 0.8 and lactate of 0.7 mmol/L. Routine venous blood tests did not identify any elevated markers of infection or inflammation. A chest radiograph (Figure 1) showed cardiomegaly and pulmonary oedema.
Figure 1
The patient was administered oxygen and given a diuretic to improve his ventilation.
The working diagnosis was congestive cardiac failure in the presence of what was presumed to be a new murmur. Urgent echocardiography revealed an aortic root of 6.2cm diameter at sinus level, with an evident dissection flap. There was no obvious haematoma. Severe free flowing aortic regurgitation, a dilated hyperdynamic left ventricle and a 0.7 cm diameter pericardial effusion anteriorly were also noted. It was concluded that the patient had a sealed 7cm type A aortic dissection. This was confirmed by a CT scan (Figure 2).
Figure 2
Large bore venous access was obtained and an intravenous beta blocker (Labetalol) administered. Urgent transfer to a tertiary cardio-thoracic surgical centre was made. He underwent aortic root and valve replacement, along with coronary artery bypass grafting to the right coronary artery using a reversed long saphenous vein graft. Postoperatively, he was anticoagulated on Warfarin, and was also placed on beta blockade therapy (Bisoprolol), a diuretic (Frusemide), an ACE inhibitor (Ramipril), and a statin (Simvastatin).
Discussion
Aortic dissection is a medical emergency. If left unrecognised or untreated mortality can be as high as 80% in two weeks, or 90% within three months1,2. 96 % of patients with aortic dissection present primarily with chest pain. The remaining 10% present with symptoms secondary to impairment of blood supply to other organ systems3. Dissections involving the ascending aorta present with retrosternal chest pain, while interscapular pain suggests involvement of the descending aorta. Pleuritic pain may indicate haemorrhage in the pericardial sac, with the potential for acute cardiac tamponade.
Only 6% of aortic dissections present with acute congestive cardiac failure. Patients presenting with aortic dissection and congestive cardiac failure are more likely to present without chest pain and have a valvular abnormality. When chest pain is present, the pain is more often mild and less likely to be abrupt in onset. Patients are less likely to be hypertensive on presentation and more likely to present in shock. These patients are more likely to have Stanford type A dissection . Congestive cardiac failure does lead to a delay in surgical intervention4.
Congestive cardiac failure is usually due to aortic regurgitation from aortic valve disease, incomplete aortic leaflet closure, or aortic valve disruption. In the setting of unexplained cardiac failure aortic dissection should be considered, especially when an aortic regurgitant murmur has been detected clinically. Heart failure has been associated with supravalvular aortic stenosis in the presence of a painless type A dissection, in a patient presenting with persistent cough5. Rupture of aortic dissection into the right atrium, right ventricle, or main pulmonary artery may lead to a left to right shunt and congestive heart failure6.
Painless aortic dissection has been recorded in other contexts, particularly with chronic dissection and in patients with Marfan’s syndrome.The absence of chest pain should not exclude aortic dissection.
‘Munchausen Syndrome by Proxy’ (MSBP) was first described by Meadow in 1977 1, as a form of child abuse in which the caregiver (generally the mother) causes or simulates illness in the child for psychological gain. Three features are required for diagnosis: 1. The history, signs and symptoms of disease are not credible; 2.The child is receiving unnecessary and harmful, or potentially harmful, medical care; 3. If so, caregivers are instrumental in instigating the evaluations and treatment 2. As has been emphasized, the motivation of the mother (her psychological needs), is important in distinguishing MSBP from other forms of Paediatric Condition Falsification (PCF) 3. Initial reports described young children with acute, life-threatening events, such as sleep apnoea, epilepsy, diarrhoea, or bowel obstruction 4,5. Sudden Infant Death Syndrome (SIDS) has an ominous association with MSBS which has survived controversy 6-9. MSBP has been reported in children in learning disability settings;10 whereas there have been few, if any, reports in general child psychiatry. Causal factors for this may be that it is perceived to be less exciting, of a multi-disciplinary nature, and, arguably, due to more humdrum circumstances which surround it. Initial descriptions, indeed, required that the child present with organic symptoms 11. Psychiatric presentations may be more difficult to detect because of the paucity of objective diagnostic tests, with the consequent greater reliance on the history provided by caregivers. Caregivers who simulate school refusal in their children have been considered not to have MSB 12, (though they do show PCF). The exclusion of psychiatric presentations is noteworthy, because such psychiatric presentations may differ in important ways from the typical, dramatic, presentations seen in very young children. Since psychiatric diagnosis in children is so dependent upon parental history, such cases may also be more difficult to detect, and therefore more common than previously thought. Three cases are described here, which, though typical of MSBP in many ways, differ in their psychiatric symptoms, in their age, and in the prominence of school refusal.
Cases
Case X
This girl was born at 37 weeks gestation by emergency caesarean section because of a slowing of the foetal heart rate. She was the eldest of 4 children. Her father was severely disabled with a chronic degenerative disease. No distress was evident at birth, but she had short stature (3rd centile for height and weight). At 9 years of age, she presented to a Child & Adolescent Mental Health service with low self-esteem and an eating problem. Her parents attributed this to bullying at school, which, the mother told doctors in a subsequent interview, took place when the child was 7, and was characterised by beatings so severe as to leave her with multiple bruises, and ‘a voice which changed following this trauma’.
The girl was found to have an IQ in the borderline range. Her mother reported that she had 'allergies to milk and eggs'. At another interview she said the allergy was to ‘shellfish and nuts’, and that it had been discovered when she developed an anaphylactic reaction in another country at 18 months of age. The mother also claimed that eczema was diagnosed at that time. Her diet was restricted, and her mother commented that she 'needed to buy special foods', and that 'asthma developed at age 7'. The child was found to be unhappy, but not depressed. There was no evidence of weight loss, but an eating disorder was diagnosed on the basis of maternal reports.
X’s brother had been diagnosed as having ADHD, and was prescribed psychostimulants. Aggression and odd behaviour had given rise to suspicion of an autism spectrum disorder. After a number of assessments, no diagnosis was made. Compliance with medication was a problem, the mother claiming that, although she personally administered tablets, he had somehow avoided swallowing them, and adduced this as a reason for their lack of efficacy. The mother later reported that 'he was doing extremely well off all treatment'.
The mother expressed dissatisfaction with the attention and care her daughter had received from one service. However, a letter, written shortly afterwards, gives an insight into her personality, and the relationship which had developed with professionals. She wrote: ‘I did not realise I was so overpowering towards you all... I only feel constant pain & hurt at what has happened to our daughter. Let me know if you do not want to see us anymore.’
It was at this point that the ability of the parents to care for their children was questioned, when it was discovered that the mother had been leaving the children unsupervised overnight, so that she could help with a scout camp (with which none of her own children were involved). School authorities were also alarmed that the children were left unsupervised for long periods before school opening, and that they arrived hungry and dishevelled. The mother opposed involvement by social services, and, in the absence of any formal complaint, they were not notified.
Unhappy with the treatment she received from her psychiatrist, the mother sought a second opinion. To this doctor, the mother reported that there had been an increase in frequency of nocturnal enuresis at 7 years. She reported that her daughter was happy at school, but that there were problems at home. She reported that her daughter was 'unable to swallow solid food'; she was 'able to take liquidised food only', and 'she became increasingly anxious about solids'. The mother said she had contacted the Anorexia Nervosa Society ‘who advised she had a phobia about food'. The mother reported that when she gradually re-introduced solids, eating recovered and that appetite returned.
At 14 years, X’s mother told a paediatrician that her problems started at age 7, when she had been bullied by several children in the class. This doctor found her to have constitutional short stature. No allergies were reported or found. The mother resisted attempts to obtain copies of previous medical records. A neurologist found 'no evidence of regression'. The mother quoted a paediatrician to another professional as having said, 'It would be disastrous for her if she had a period', and she told another paediatrician that the referring doctor had said she was being referred, 'to sort out her psychological problems immediately’. She was referred to a third child psychiatrist, who found low mood, social withdrawal, and 'recent adjustment problems'. She was then referred again to local psychiatric services for follow-up. At this stage, her mother wrote: '... it is now confirmed that her short stature has a psychological basis, and that this started at 6 years of age because of bullying'.
At her mother’s insistence, X was seen then by another psychiatrist, ‘to discover the psychological cause of her short stature’. No psychopathology was found. Mother declared that she was ‘astounded’ and demanded to know how the doctor could explain ‘the dramatic fall in IQ which had taken place at age 10’.
By the time of referral to social services, the child had been seen by 3 paediatricians, 4 psychiatrists, and numerous other mental health professionals.
Case Y
This boy was born without complication, developing normally up to middle childhood. When he was 3 years old, a younger sister was born. She died at the age of 3 months, and a coroner's verdict of SIDS was returned. His mother experienced a pathological grief reaction.
The family holidayed abroad periodically; such trips were frequently marked by attendance at the A&E department of the local hospital, where the boy presented with sundry somatic complaints. There were occasional brief hospitalisations, but no physical abnormality was ever diagnosed.
The child’s mother made several unsubstantiated allegations of bullying at school. His father disputed these, though he did not oppose them. Thorough investigation failed to substantiate any such episode. The father complained to doctors of his powerlessness in the face of his wife's over-protective, domineering approach; he recognised the harmful effect this was having on their son.
Whereas the mother complained that her son had 'loss of interest', 'poor concentration', was ‘not eating', and had suffered weight loss; the boy told doctors, 'I'm good at school'; and 'I'm always happy with my friends'.
His mother had a benign brain tumour, which had presented initially with epilepsy when she was in her teens, but was well controlled. During her mid-40’s, she presented frequently with pseudo-seizures and other odd neurological symptoms. To each doctor she met, she presented herself has having a 'terminal brain tumour'. She told her neurologist that she was 'distancing herself from her son’, as she was ‘going to die soon’, and ‘he would have to be tough enough to get on without her'.
On one occasion, when brought to hospital because of suicidal ideation, Y commented that ‘school was good’, and that he had ‘a few good friends’. He listed a few favourite subjects, and responded appropriately to wishes about the future. The conclusion was that he was euthymic. Notwithstanding his reports to doctors, he was kept at home for prolonged periods because of 'bullying'. The mother claimed that her husband 'can be abusive to the child when angry', though the impression of numerous psychiatrists was that he had a passive, dependent personality. He begged professionals for help in protecting his child, and when a referral to social services was eventually made, he was profuse in his gratitude.
Case Z
This boy's monozygotic twin brother presented to a private psychiatrist at 1 ½ years of age with 'behavioural problems', and a diagnosis of autism was made. No standard observational assessments were performed, nor neurodevelopmental history taken. He was referred to specialist autism services, who found no abnormality following a multi-disciplinary assessment. When the twins were 7 years of age, the mother brought the other twin, Z, to community child psychiatric services with vague concerns regarding 'development' and 'social skills'. By this she meant that he '[had eaten] his food off the floor when he was younger', and that he currently 'behaved in a silly, immature way'. She said that 'teachers complained of disruption', and she, in turn, complained of poor cooperation from the school. Independent contact with the school disclosed no such behavioural concerns. Her husband spent most nights away on business, and the twins slept in her bed. They 'would not settle for hours' and she 'had to sing to get them to sleep'.
His mother had been treated for depression in the past. She reported that she had given up her job in order to look after her children, whom she felt had 'special needs'.
At clinical interview she was extremely reluctant to leave her son, and her prolonged departure from the room was accompanied by exaggerated displays of affection, which clearly embarrassed her son. He, on the other hand, separated easily. On his own, he was initially reserved, but became talkative and happy when discussing his friends, school, interests, etc. There was no evidence of any autistic-type disorder, nor indeed any other psychiatric problem.
At the time of writing, she has refused to accept the assurances of two different services that there was nothing wrong with her son, and she was continuing with attempts to have him re-assessed.
Discussion
All three of these children presented with complaints from mothers, which, while perhaps credible in isolation, became more and more far-fetched when viewed together as a whole. In all cases harm resulted to the child from excessive investigation, social isolation, and absence from school. In all cases, mothers had narcissistic personality problems, and fathers had a subordinate role (one because of chronic illness, another because of a dependent personality). All children had siblings with dubious or unexplained illness. All cases involved many physicians and allied professionals, who became involved in what transpired to be, when they started to communicate with one another, a pattern of simulated illnesses and symptoms. There was a general reluctance to refer to social services, and a delay in their involvement, perhaps because of the absence of acute or marked abuse, and because the caregivers seemed the opposite of the 'typical' negligent, abusive mother. These children were somewhat older than most cases of factitious disorder by proxy, who also differ in presenting with acute, life-threatening events or surgical emergencies. Although probably less lethal, psychiatric presentations may offer more scope for abuse, due to the greater reliance on parental reports in child psychiatry. Unwitting collusion by schools, in part caused by an understandable sensitivity to bullying allegations, may have facilitated presentation to psychiatric services.
Most cases of MSBP have emerged from the U.S. The phrase ‘Psychological needs’ has been emphasised, and suggests vagueness, an impression strengthened by the initial psychodynamic terms in which these case were couched. It may be a useful term, nonetheless, in that it distinguishes motivations such as revenge, delusions, or poverty, from those in which the perpetrator behaves in this way to, for example, fool doctors or exhibit herself as an ideal parent 13. Schreier and Libow 14 suggested that a key psychological factor may be an ingratiating relationship which the mother pursues with the child's physician, who tends to be male, isolated, and idealistic. The preponderance of mothers among perpetrators may be due to a satisfaction obtained by deceiving ‘authority’ or ‘power’ figures, but this (and any other explanation) has not yet been substantiated. Such manipulation was not present in any of these three cases. This may be due to the central role which the family doctor retains in many other countries, and to the multi-disciplinary nature of health care, particularly mental health services, in other health systems, which precludes the doctor from seeming a ‘hero figure’.
‘Munchausen Syndrome by Proxy’ is clearly not a disease, and can be considered a disorder only by analogy. This, as well as the general tendency in medicine to abandon eponymous diagnostic labels, argues for use of the term ‘Paediatric Condition Falsification’, preferred by the American Professional Society on the Abuse of Children (APSAC) 15; or ‘Factitious Disorder by Proxy’, as preferred by the Diagnostic & Statistical Manual of Mental Disorders (DSM) 16. Notwithstanding these compelling reasons, the radically different profile of the ‘caring mother’, in cases like those under discussion, make it essential to distinguish the situation described in this case series from other circumstances in which mothers harm children. The mother’s motivation is crucial if the child is to be protected: specific remedies in other circumstances may offer hope of an amelioration, but the rate of recidivism in ‘Munchausen Syndrome by Proxy’ 17,18, means that these children will require vigilance and protection for as long as they are in contact with their mother.
It is important to consider how these episodes might have been detected earlier. Good history-taking would have revealed the falsehood of an allergy to dairy products in the first case. A higher index of suspicion might have led to greater communication, and better detection, among disparate professionals as in the case of Munchausen Syndrome in adults. Formal channels for reporting concerns without fear of recrimination could be established in hospitals and out-patient settings. Greater institutional support for healthcare workers with concerns, as well as broader awareness among family doctors, nurses, psychologists, and social workers, is a prerequisite. In medical presentations, a bizarre, inconsistent history, a failure to cooperate with attempts to obtain medical records, features of a maternal histrionic or narcissistic personality, and any history of abuse towards other siblings, should raise the alarm. These apply also in the case of psychiatric presentations. In medical presentations, abuse can be detected by observing ‘recovery’ of the child when removed from the parent’s reach. The chronicity and gradual nature of psychiatric symptoms make these cases appear less dramatic, and such a ‘test’ impractical, but certain other features may help. Unconvincing reports of bullying at school, despite thorough investigation, poor school attendance without an adequate explanation, and an incongruity between maternal reports and the child’s mental state, may all be helpful.
Conclusion
Reports of Psychiatric presentations of Paediatric Symptom Falsification are rare, but there are good reasons for suspecting that the true incidence may be higher. Psychiatric presentations are probably not typical of Paediatric Symptom Falsification, and may for this reason be missed. Experience shows that reporting to child protection agencies is essential, but this is difficult and tends to be avoided for various reasons. There is a need for education of mental health and allied professionals in this condition so that much suffering of children can be avoided. Paediatric Symptom Falsification involves psychological problems in the caregiver (generally the mother), which are very difficult to manage. These require understanding and compassion, but should not be a barrier to protecting the child.
The authors declare that they have no conflict of interest.
The legal high ‘Ivory Wave’, also known as ‘Ivory Coast’, ‘Purple Wave’ or ‘Vanilla Sky’, is a designer drug that has become popular among clubbers in the United Kingdom (UK) after mephedrone was banned in April 2010.1 Ivory Wave is advertised as a relaxing bath salt and has been freely available on the Internet for about £15 a packet (200mg).2 Three different versions have been on the market, namely, Ivory Wave, Ivory Wave Ultra (also known as Ivory Wave 2), and Ivory Wave 3, although their differences are unknown.3 Studies have shown that Ivory Wave contains cathinone-derived stimulants and, when snorted in high doses, bring similar effects to those of amphetamine and ecstasy.4
Recently, clusters of hospital admissions have been reported around the UK following the use of Ivory Wave. The majority of patients were described to have ‘acute paranoid psychosis’ with severe agitation, which wore off after a couple of days.5 However, some patients had more serious physical complications and had to be monitored in the coronary care units for up to 12 hours.2, 5
Following the increase in the number of Accident and Emergency (A&E) admissions relating to Ivory Wave, healthcare professionals have expressed their concerns about the harmful effects of the substance. The Department of Health has issued advice on handling the users who may present to health services for help.6 However, the literature is limited on the physical and psychological effects of the substance at present. Therefore, we report our case here to describe some of the clinical features of Ivory Wave misuse.
Case presentation
A 26-year-old Caucasian male, with a background history of obsessive-compulsive disorder (OCD) and depression, attended an Accident A&E after snorting approximately 700mg of ‘Ivory Wave Version 3’ in a day. He presented with severe agitation, persecutory delusions, and auditory and visual hallucinations. He stated that ‘people’ were trying to kill him and his mother with a knife, and he could hear their voices threatening to kill him. He also complained of mild/moderate breathing difficulty and involuntary movements of his arms and feet.
In recent years he had been ‘experimenting’ with several legal highs, including Ivory Wave, ‘Charge’ and ‘Mojo’. Five weeks prior to this admission he had visited A&E with a similar presentation, but without persecutory delusions, after sniffing an unknown amount of ‘Ivory Wave 2’. The hallucinations shortly disappeared and he was discharged home.
Otherwise, he was physically fit and well. He had a long history of severe OCD with borderline psychotic features/social anxiety where he was consistently worried about what other people may do to him. There was no personal or family history of psychosis. He was taking clomipramine (125mg) and olanzapine (12.5mg) for OCD and depression but his compliance had been erratic before the admission.
In the present admission he was very agitated and restless. He had non-goal-directed involuntary movements on both arms and feet: repetitive flexion of his elbows and dorsiflexion of his ankles. The physical examination showed that he was pyrexial with a temperature of 37.9°C and had bilaterally dilated pupils. The respiratory examination was normal with an oxygen saturation of 98% on air. The heart rate was slightly fast at 109 beats per minute (bpm) and blood pressure was 122/82 mmHg. The rest of examination was unremarkable with a normal electrocardiogram (ECG). Laboratory investigations revealed a raised white blood cell (WBC) count of 23.5 ´ 109/L and C-reactive protein (CRP) of 332 mg/L. He also had hyponatraemia (Na+ 126 mmol/L) and elevated creatinine kinase (CK = 662 iu/L). Urine drug screen was negative to amphetamine, opiates, cannabinoids and cocaine.
Initially the patient was admitted to a medical ward and commenced on normal saline with intravenous antibiotics (co-amoxiclav) because of the raised inflammatory markers. The body temperature, CRP, and WBC count fell gradually; the CK level dropped as well. The blood culture came back negative. However, the patient remained agitated, was running around the ward, and experiencing visual and auditory hallucinations. He required PRN lorazepam and regular diazepam.
On day five of admission the patient was deemed medically fit and discharged from the medical ward. However, he was still agitated and confused about what had happened. Concerns were raised regarding his mental state, given his past psychiatric history and current problems. A Mental Health Act assessment was performed and the patient was admitted to a psychiatric unit under Section 2 of Mental Health Act 1983 on the same day.
On admission to the unit the persecutory delusions and hallucinations were still present but to a mild degree. The involuntary movements appeared more like muscle twitches, which occurred less frequently. He was observed on the ward and only PRN lorazepam was prescribed together with his regular medication. He then settled on the ward and did not require any further PRN medication.
After a few days the persecutory delusions and hallucinations wore off. The involuntary movements had stopped but left patches of numbness on his right arm, mainly on his fingers. The area was poorly defined and was not localized to a specific dermatome. The numbness disappeared after a few days without any complications.
The patient remained in the psychiatric unit for two weeks before discharge to the care of the Community Mental Health Team (CMHT).
Discussion
The exact components of Ivory Wave are unclear and thought to be variable.4 , 7 Studies have shown that the main ingredients include MDPV (3,4-methylenedioxypyrovalerone); desoxypipradrol, also known as 2-diphenylmethylpiperidine (2-DPMP); and lidocaine.7, 8. 9 MDPV and desoxypipradrol are both synthetic stimulants. MDPV was first synthesized in 196910 and is found as a white or light tan powder.4 Desoxypipradrol was initially developed by a pharmaceutical company in the 1950s as a treatment for Attention Deficit Hyperactivity Disorder (ADHD) and narcolepsy, but it was replaced by other related substances.8 They both act as noradrenaline and dopamine reuptake inhibitors and their effects are thought to be similar to those of amphetamine and cocaine in high doses.9, 11, 12
Ivory Wave is known to bring on several desired effects, including increased energy and sociability, increased concentration, and sexual stimulation. There are also many physical and psychological unwanted effects reported including insomnia, severe agitation/anxiety, panic attacks, kidney pain, stomach cramps, tachycardia, hypertension, dilated pupils, headache, tinnitus, skin prickles and numbness, dizziness, and dyspnoea.4, 13 These effects appear highly dose-dependent4 and have been based on self-reports made by users on online forums.
In the UK there have been several media reports of hospital admissions related to Ivory Wave. The majority of patients were described to have acute psychotic symptoms, namely paranoid delusions, and auditory and/or visual hallucinations. A few of them had physical complications, requiring cardiac monitoring in ICU.1, 2, 5 However, no detailed description of their clinical features were available.
In this case report, we have described chronologically the clinical features of a patient, who presented to A&E after taking Ivory Wave. The patient had a similar presentation to what it was described by Paolo Deluca and his colleagues.4 The patient also experienced involuntary movements in his limbs which has not been reported before in the literature. We have also reported the blood test results: raised inflammatory markers (WCC and CRP) and CK.
The findings from this case, in combination with the limited literature, suggest that the use of ‘Ivory Wave’ can lead to serious complications including over-stimulation of the cardiovascular and nervous system, hyperthermia, and acute psychosis which can potentially result in severe illnesses or even death. The risk of these effects would be greater if the drug was combined with other recreational drugs or alcohol. In addition, the exact composition and strength of the substance may vary and users may not be completely aware of what chemicals they are consuming. This implies that users of Ivory Wave may to taking potentially dangerous substances with unknown effects.
In April 2010, MDPV was made a Class B drug in the UK together with other cathinone derivatives. In addition the UK Home Office has recently announced a ban on the import of desoxypipradrol and any products containing the chemical.15 The use and availability of Ivory Wave in the UK is being closely monitored and may result in further legislative review. Changes in legislation, more research studies, and health education on Ivory Wave could help the public to realize that, irrespective of the legal status of a drug, recreational use of substances may pose a significant risk to their health.
Introduction A hydatid cyst is the larval stage of a small tapeworm, Echinococcus granulosus. This is an emerging zoonotic parasitic disease throughout the world, thought to cause an annual loss of US $193,529,740.1 Hydatid cysts are more prevalent in Australia, New Zealand, South America, Russia, France, China, India, the Middle East and Mediterranean countries.2,3,4 They are most commonly (about 50-75%) seen in children and young adults.4,5,6 The liver is the most common organ involved (77%), followed by the lungs (43%).7,8,9,10 However, some researchers report that the lung is the most common organ involved in children, possibly due to bypass of the liver by lymphatics, and higher incidental findings in the lungs when children are assessed for other respiratory infections.8,11,12,13 Hydatid cysts have been reported in the brain (2%),3,4,5,7,8,14,15 heart (2%),8,10,13,16 kidneys (2%),9,10,11 orbit (1%),17,18 spinal cord (1%),3,19 spleen,4 spine,3,8 spermatic cord20 and soft tissues.8 However, in the Mediterranean region, the incidence of brain hydatid cysts have been reported higher (7.4-8.8%).21 Surgery remains the treatment of choice, although recently some new modalities have been described.5,8,22 Careful removal of the lesion is of considerable importance, otherwise fatal complications are inevitable.23,24,25 We describe the case of a 6 year old boy who came to our department with various neurological manifestations. The main purpose of this study is to demonstrate the unusual symptoms of the patient and the enormity of the operated cyst, which was fully resected without rupture. Case Report A 6-year-old boy was referred to our Neurosurgery Department with a four week history of ataxia and left sided weakness. His vital signs were normal and his Glasgow Coma Scale (GCS) was 15. The symptoms had started about six months ago with numbness and parasthesia of the toes. Subsequently he developed intermittent nausea and vomiting. He then started to develop left sided weakness and finally ataxia. He also had a few focal convulsions but did not complain of headache. Fundoscopy revealed bilateral frank papilloedema. On examination, the patient had nystagmus and a positive Romberg’s test. Laboratory data showed mild leucocytosis without any significant rise in eosinophils, and liver enzymes were normal. The enzyme-linked immunosorbent assay (ELISA) for hydatid cysts was negative. Plain chest X-ray and ultrasound scan of the abdomen and pelvis were also normal. Brain computed tomography (CT scan) of the frontal and parietal lobes demonstrated a single large, spherical, well-defined, thin-walled homogenous cyst, with an inner density similar to that of cerebrospinal fluid (CSF), and a wall which did not show enhancement [fig 1(a)]. This cystic structure caused a mass effect and a midline shift towards the left, as well as hydrocephalus, possibly due to obstruction. Magnetic resonance imaging (MRI) of the brain showed cystic signal intensity similar to that of CSF, without ring enhancement or oedema [fig 2]. Fig 1 (a): Pre-operative unenhanced CT scan which shows a large CSF density cystic lesion on the right side causing mass effect and midline shift to the left. There is no peri-lesional oedema. Fig 1 (b): Post-operative CT scan of the lesion shows a large void which can lead to dangerous collapse. Mild haematoma is also seen. Fig 2 (a): T1-weighted axial MRI of the brain demonstrates a cyst density similar to CSF. Fig 2 (b): T2-weighted MRI shows no ring enhancement or oedema. The periventricular hyperintensity of the left side is probably due to obstructive hydrocephalus. Fig 3: This shows the cyst removed in toto after operation. The cyst appears creamy and smooth. After summation of all the above data, the diagnosis of a hydatid cyst was made and a right frontotemporoparietal craniotomy was performed. A large cystic structure (14×14×12 cm) was delivered with utmost care to avoid rupture and spillage [fig 3]. A hydatid cyst was confirmed by pathology reports. A post-operative CT scan showed a large space without any residual matter [fig 1(b)]. Post-operatively, albendazole 15 mg/kg was started and continued for four weeks. The patient showed marked improvement in his neurological deficit and was discharged after one week with close follow-up.Discussion/Review Of LiteratureLife CycleHydatidosis is caused by Echinococcus granulosus, which occurs mainly in dogs. Humans who act as intermediate hosts get infected incidentally by ingesting eggs from the faeces of the infected animal. The eggs hatch inside the intestines and penetrate the walls, entering blood vessels and eventually reach the liver where they may form cysts or move on towards the lungs. Even after pulmonary filter, a few still make it to the systemic circulation and can lodge in almost any part of the body, including the brain, heart and bones.2,3,8,14,16,26 Brain hydatid cysts are relatively rare and only account for up to 2% of total cases.4,5,7 The actual percentage may be higher than what we have in literature, due to under-reporting. Brain hydatid cysts can be primary (single) or secondary (multiple).2,3,4,5,7 The latter are thought to arise from the multiple scolices released from the left side of the heart following cyst rupture in the heart2,3,5,27 or due to spontaneous, traumatic or surgical rupture of a solitary cranial cyst.3,5 Cysts mostly involve the territory of the middle cerebral artery4,7 but other regions like intraventricular, posterior fossa and the orbit have also been reported.15,17,18,28 The wall of the cyst consists of an inner endocyst (germinal layer) and outer ectocyst (laminated layer). The host reacts to the cyst forming a pericyst (fibrous capsule), which provides nutrients to the parasite. In the brain, due to minimal reaction, the pericyst is very thin. The endocyst produce scolices which bud into the cyst cavity and may sediment within the hydatid cavity, commonly known as hydatid sand.3,14,29,30Presentation and DiagnosisMost hydatid cysts are acquired in childhood and are manifested during early adulthood.8,29 Cysts develop insidiously, usually being asymptomatic initially, and present with protean clinical and imaging features.3,5,6 In previous studies the most common presenting symptoms were headache and vomiting.4,5,7,14,15,28 Also in the literature, patients reported ataxia, diplopia, hemiparesis, abducens nerve palsy and even coma.5,7,15,28 Surprisingly, in the present study the patient did not have a headache and presented with parasthesia and numbness of the toes. Later he developed left sided weakness, convulsions and finally ataxia, which correlate with previous studies. Diagnosis of a hydatid cyst can sometimes be confused with other space occupying lesions of the brain, especially abscesses, neoplasms and arachnoid cysts.14,31 In this study the patient had bilateral frank papilloedema which is also mentioned in earlier reports.4,28 The Casoni and Weinberg tests, indirect haemagglutination, eosinophilia and ELISA are used in diagnosing hydatid cysts, but as brain tissue evokes minimal response many results tend to be false negatives.2,5,8,25 In our case also, serology for hydatid cyst was negative. CT scan and MRI are used frequently in diagnosing the cystic lesions.3,8,14,23,32,33 However, MRI is considered superior in demonstrating the cyst rim.5,8,11,21,32,34 On CT scan, a solitary cyst appears as well-defined, spherical, smooth, thin-walled and homogeneous, with an inner density similar to CSF, and non-enhancing walls.11,29,32The wall may appear iso-dense to hyper-dense on CT scan3,8, and rarely, may become calcified.11,29,32 There is usually no surrounding brain parenchymal oedema, which if exists along with ring enhancement, indicates inflammation and infection. 7,11,32,33,34,35 Ring enhancement and peri-lesional oedema differentiates brain abscesses and cystic neoplasms from uncomplicated hydatid cysts.3,8 These findings can in fact sometimes cause dilemma and misdiagnosis and lead to catastrophic events.14 The cyst shows low signal intensity on T1-weighted, and high signal intensity on T2-weighted MRI.2 MRI may also show peri-lesional oedema not seen on regular CT scan imaging.7 MRI may prove superior in determining exact cyst location, presence of super-added infections and cystic contents, and also in surgical planning and ruling out other diagnostic possibilities.14,33 We strongly recommend MRI for better evaluation of cystic brain lesions. Spontaneous cystic rupture can lead to different appearances depending on which layers have been obliterated, and produce some specific signs.3 When only the endocyst ruptures, cyst contents are held by the outer pericyst giving a peculiar water lily sign, which is pathognomic.3,8TreatmentThough still in infancy, medical therapy for small or inoperable brain hydatid cysts has been promising. Albendazole alone or in combination with other compounds, such as praziquantel, has been reported with favourable results as an adjunct and, in certain circumstances, as the primary mode of treatment.2,36,37,38 It is reported that albendazole results in the disappearance of up to 48% of cysts and a substantial reduction in size of the cysts in another 28%.2 The duration of the treatment is four weeks or more, and recently many authors have favoured a prolonged therapy. The change in levels of cyst markers such as alanine, succinate, acetate and lactate, measured before and during treatment on Proton Magnetic Resonance Spectroscopy (MRS), correlate well with shrinkage and resolution of cyst findings on conventional MRI and help in evaluating the efficacy of chemotherapy.39 Cysts may drain into ventricles or rupture completely, causing spillage of contents into the subarachnoid space, leading to fatal anaphylactic shock, meningitis or local recurrence.3,5,22,25 Surgery is the mainstay for treating intracranial hydatid cysts and the aim is to excise the cysts entirely without rupture, which can otherwise lead to catastrophic events as described earlier 2,3,14,25. The Dowling-Orlando technique remains the preferred method, in which the cyst can be delivered by lowering the head of the operating table and instilling warm saline between the cyst and the surrounding brain.40 Even minimal spillage can cause deleterious effects (1 ml of hydatid sand contains 400,000 scolices).14 The thin cyst wall, periventricular location and micro-adhesions to the parenchyma are the main problems encountered during the surgical procedure.1,22 The large cavity remaining after the cystic removal can lead to many serious complications, such as cortical collapse, hyperpyrexia, brain oedema and cardio-respiratory failure.5 Recurrence remains a major concern, which is managed by both antihelminthic chemotherapy and surgery. In a study conducted by Ciurea et al, 25% of the patients had recurrence, which highlights the need for long term follow up.23 In the present study, due to the huge size of the cyst and progressive neurological deficit, it was not wise to completely rely on medical therapy. Surgery was performed and post-operatively albendazole was started as an adjunct. We recommend that for treating brain hydatid cyst, the size of the cyst, multiplicity, location and neurological deficit must all be taken into consideration.
Liver abscess accounts for 48% of visceral abscesses 1 and presents with significant morbidity and mortality. The overall incidence of pyogenic liver abscess is 3.6 per 100,000 populations, 2 however; elevated pancreatic enzymes within the content of a liver abscess have never been reported in the literature.
CASE REPORT:
A 36-year-old African American male with a history of chronic pancreatitis presented to the emergency department for abdominal pain in the epigastric area along with nausea, vomiting, diarrhoea, fever. His symptoms began 3-4 days before presentation. The abdominal pain was dull in nature and 6/10 in intensity, non-radiating. His past medical history was significant for HTN, diabetes mellitus and chronic diarrhoea secondary to chronic pancreatitis.
On admission the patient was alert and oriented, blood pressure was 97/44 mm Hg, heart rate 16 beats per minute, respiration 16 per minute, oxygen saturation 94% on room air and temperature 102*F. Abdominal examination revealed hyperactive bowel sounds and tenderness in the epigastrium & RUQ. Liver span was 14 cm. The rest of the examination was unremarkable.
Laboratory work revealed: Haemoglobin 9.8 g/dl, WBC 22.1 Thou/mm3 with segmented neutrophils of 81% and 9% bands, BUN 54 mg/dl, Cr 4.7 mg/dl, total protein 10.4 g/dl, albumin 1.8 g/dl, total bilirubin 1.1 mg/dl, direct bilirubin 0.3 mg/dl, AST 98 IU/L, ALT 38 IU/L, alkaline phosphatase 250 IU/L, amylase 81 units/L, lipase 10 units/L, lactate 2.3 mmol/L and INR 1.39.
The patient was started on fluids and meropenem for broad spectrum coverage. However his condition worsened and he developed acute respiratory distress syndrome secondary to sepsis necessitating intubation. Due to his abdominal pain he underwent a computer tomography (CT) scan of the abdomen, which revealed pancreatic calcifications and multiple liver abscesses; the largest measuring 7.5cm in the right lobe of the liver (Figure 1).
Figure 1
As the patient’s condition did not improve, he underwent liver abscess drainage. Fluid analysis showed ph 4.0, LDH 39 units/L, glucose 81 mg/dl, protein 1.6 g/dl, lipase of 16 units/L and amylase 68 units/L. The presence of amylase and lipase in the liver abscess without any evidence of a fistula between liver and pancreas on CT scan was unexpected, therefore it was decided to leave the catheter in situ for continuous drainage. 3 Even though his blood and fluid cultures remained negative during the hospital stay he was continued on antibiotics, which may have meant that the initial antibiotic therapy rendered the blood cultures negative. The success of management was assessed with a hepatic CT 10 days post drainage and was demonstrated by the observation of improvement in the patient’s general condition, as indicated by normal temperature, decreased draining catheter output and the resolution of deranged laboratory values. The catheter was then removed and the patient was discharged.
DISCUSSION:
Liver abscesses develop via seeding through portal circulation, directly via spread from biliary infections or from surgical or penetrating wounds and also from systemic organs via haematogenous spread. In our case the most reasonable explanation was through the involvement of portal circulation due to recurrent pancreatitis.
The morbidity and mortality rate for liver abscesses ranges from 2 – 12 % depending on the severity of underlying co-morbidities. 2 The clinical manifestations, as in our case, are characterized by abdominal pain (50-75%), 4, 5 fever (90%), nausea and vomiting. Other symptoms may include weight loss, malaise and diarrhoea. On physical exam RUQ tenderness, guarding, rebound tenderness, hepatomegaly and occasional jaundice can be appreciated. The diagnosis of a liver abscess can be made by radiographic imaging followed by aspiration and culture of the abscess material. Liver abscesses can be either polymicrobial or monomicrobial, unlike in the case with our patient, whose abscess was sterile. Depending on the microbial results additional sources of infection should be evaluated. Drainage of abscesses can be percutaneous 6 or open surgical. Percutaneous drainage with coverage of antibiotics was successful in our patient.
CONCLUSION:
In summary, we present a case of pancreato-liver abscess in a patient with a history of chronic calcified pancreatitis. It was treated with antibiotics and percutaneous drainage, with satisfactory resolution. To our knowledge this has never been reported in the literature and more work needs to be done to understand the pathophysiology of elevated pancreatic enzymes in the context of a liver abscess in a patient with a history of chronic pancreatitis.
Myocardial ischemia from coronary artery vasospasm can lead to variety of presentation including stable angina, unstable angina, myocardial infarction and sudden death 1. Although, pathognomic clinical scenario includes symptom of chest pain, transient ST-segment elevation on the electrocardiogram(ECG), and vasospasm on a coronary angiography, atypical presentations have also been reported 2. Various known physiological factors including stress, cold, hyperventilation and pharmacological agents including cocaine, ethanol, 5-Fluouracil, and triptans can precipitate a vasospastic attack. 3-7.We report a case of ST-segment elevation due to right coronary artery vasospasm, in patient with hypoxic respiratory failure, and successful treatment with calcium channel blockers.
Case description
A 56 year old man was admitted for the repair of a large ventral incisional hernia. The patient had a prior history of morbid obesity, chronic obstructive pulmonary disease (COPD), hypertension and cigarette smoking. The postoperative course was complicated by bilateral pneumonia leading to respiratory failure requiring mechanical ventilation. An electrocardiogram at the time of intubation was essentially normal. Aside from bilateral rhonchi and crackles on lung auscultation, the rest of the physical examination findings were unremarkable. Arterial blood gases at the time of intubation demonstrated PH 7.33, PO2 58 mmHg, PCO2 65 mmHg, HCO3ˉ 20 mmol/L, suggestive of hypoxia and concomitant respiratory acidosis. Baseline laboratory studies including cardiac enzymes were within normal limits. The patient was treated with intravenous vancomycin for methicillin-resistant staphylococcus pneumonia. On postoperative day 4, the patient had recurrent episodes of transient ST-elevation on a bedside monitor (Fig.1).
Figure 1
These episodes lasted for 3-5 minutes and were associated with significant bradycardia and hypotension. In view of recurrent episodes, haemodynamic instability, and underlying risk factors of coronary artery disease, cardiac catheterization was performed. Coronary angiography revealed a 90% stenosis with haziness of the mid-right coronary artery without any other significant epicardial disease. An intravascular ultrasound (IVUS) was performed and was followed by administration of 100 mcg of intracoronary nitroglycerin; the lesion was reduced to almost 20%. (Fig.2).
Figure 2
The diagnosis of prinzmetals angina was made, based on clinical course and angiographic results and prompt therapy with diltiazem (120 mg per day) was initiated. The patient had no further recurrences of similar episodes during the hospitalization and on follow up at 3 months.
Discussion
The prevalence of vasospasm has been reported to be higher in Japanese and Korean population as compared to the western population. A recent multi-institute survey in Japan documented spasm in 921 (40.9%) of the 2251 consecutive patients who underwent angiography for angina pectoris 8. In contrast to the traditional risk factors for atherosclerotic coronary artery disease, the incidence of smoking, age and dyslipdaemia has been reported higher in patients with coronary vasospasm 9. Endothelial dysfunction is now considered to the major inciting factor in the pathogenesis of the vasospasm 10. Vasospastic angina (VA) with normal coronary arteries on the angiography, impaired endothelial-dependent and endothelial-independent vasodilatation has been frequently observed in these patients. Vascular tone is normally regulated by production of vasodilator factors like nitrous oxide (NO), prostacyclin and vasoconstricting agents like endthelin-1. In the presence of dysfunctional endothelium the agents that normally cause vasodilatation lead to paradoxical vasoconstriction, due to direct muscle stimulation, like acetylcholine.
Stress, whether physical or mental stress has been shown to induce coronary vasospasm and myocardial ischemia. In a study by Kim et al, coronary spastic angina was diagnosed in 292 patients out of 672 coronary spasm provocation tests. Among 292 patients, 21 (7.2%) had myocardial infarction and 14 out of these 21 had experienced severe emotional stress before the event 11. Recently, animal studies have also shown that high circulatory level of stress hormones (cortisol) exaggerate coronary vasoconstriction through Rho-Kinase activation 12. Hypoxia has been seen in animal models to predispose to vasospasm through superoxide formation, which leads to loss of vasodilator function of NO.(13)
The ECG changes that occur during attack include ST-segment elevation, and or peaking of T wave from total or subtotal coronary occlusion 1. In some cases spasm can involve more than one artery leading to ST-segment elevation in multiple leads, which may predispose to ventricular tachycardia or fibrillation 14. Coronary spasm is diagnosed by angiography, and spasm can occur at the site of atherosclerotic plaque or in normal segment of the coronary artery. In patients with equivocal diagnosis, provocative tests including administration of acetylcholine, hyperventilation to induce spasm may be required for the diagnosis.
Current first line therapy involves used of calcium channel blockers (CCB) alone or in combination with long acting nitrates. In a study comparing the effect of long acting nitrates (Isosorbide dinitrate 40mg/day) versus calcium channel blockers (amlodipine 5mg/day or long acting nifedipine 20mg/day) on coronary endothelium and vasoconstriction between patients with normal or minimally diseased coronary artery, treatment with long acting nitrates was associated with less favourable effects on coronary endothelial functions 15. Sudden withdrawal of CCB in patients with known vasospasm can lead to rebound of symptoms and may prove dangerous. In patients with refractory symptoms alpha-blockers, nicorandil have been used. Although beta blockers are believed to enhance vasospasm, Betaxalol, a selective beta-1 blocker, has been found to be effective in the treatment of variant angina due to its vasorelaxing effects 16. In addition, elimination of or control of all other risk factors or precipitants is very important for successful treatment. In drug refractory cases the percutaneous coronary intervention or coronary artery bypass graft has been performed for the ischemia relief 17.
Our patient had multiple precipitating factors for vasospasm. Endothelial dysfunction from severe physical illness and sepsis could have precipitated the VA. Furthermore, hypoxia from respiratory failure could have also been an inciting agent and cannot be ruled out. It is worth mentioning that intensive care unit patients frequently have coexistence of both the underlying risk factors and the precipitating factors for vasospasm, yet VA as a clinical syndrome is uncommonly seen or reported.
Conclusion:
The clinician needs to be aware of coronary artery vasospasm as it can pose a serious medical threat. Early diagnosis and treatment may result in improved outcomes from vasospastic angina.
A 87-year old man was referred to hospital with a five day history of lethargy and increased urinary frequency. He denied symptoms of gastrointestinal bleeding or abdominal pain. His past medical history included diabetes mellitus, chronic kidney disease, peripheral vascular disease and surgery for repair of ruptured aortic aneurysm 6 weeks ago. Systemic examination, including per rectal examination, was normal. Haemoglobin was 83g/L and C-reactive protein was 148 (Normal <5). Twelve hours after admission he developed pyrexia (37.8 degree) accompanied with tachycardia (103 beats per minute) and hypotension (BP 87/43). Soon afterwards, he had a small amount (<50 mls) of fresh haemetemesis. He also complained of lower back pain and clinical examination revealed tenderness in the left iliac fossa. He was cross-matched for blood and initiated on intra-venous fluids. As his Rockall score was six an urgent oesophago-gastro-duodenoscopy (OGD) was planned. Over the next few hours he complained of increasing central abdominal pain and had several episodes of melaena. In view of the history of recent aortic surgery and current GI bleed the possibility of aorto-enteric fistula (AEF) was considered. An urgent contrast CT scan of the abdomen (Figure 1) was therefore arranged prior to OGD.
Figure 1: Contrast CT scan demonstrating the aorta (A) with extravasation of contrast (B) and a large collection (C) around it with trapped air suggestive of infection.
Contrast computed tomogram (CT) scan of the abdomen revealed an inflammatory soft tissue mass anterior to the infra-renal aortic graft with pockets of gas and leakage of contrast into it. These findings were suggestive of an AEF. The patient was informed of the diagnosis of AEF and the need for emergency surgical repair to which he consented. During the operation the vascular surgeons found that the duodenum was adherent to the aortic graft with evidence of fistulisation and infection, thus confirming the diagnosis. Although operative repair appeared to be successful, the patient continued to bleed on the table due to disseminated intravascular coagulation and died twenty fours after admission.
Discussion
AEF is defined as a communication between the aorta and the GI tract.1 The diagnosis of AEF should be considered in every patient with a GI bleed and a past history of aortic surgery.2 Our case patient had had emergency repair of a ruptured aortic aneurysm with a prosthetic graft 6 weeks prior to his current admission.
AEFs are a rare cause of gastro-intestinal (GI) hemorrhage. AEFs can be primary or secondary. Primary AEF (PAEF) is a communication between the native aorta and the GI tract.1 The incidence of PAEF ranges from 0.04 to 0.07%.3 PAEFs commonly arise from an abdominal aortic aneurysm of which 85% are atherosclerotic.1
Secondary AEFs (SAEF) are an uncommon complication of abdominal aortic reconstruction.4The incidence of SAEF ranges from 0.6% - 4%.5 Generally two types of SAEFs have been described. Type 1, termed as true AEF develops between the proximal aortic suture and the bowel wall. These usually present with massive upper GI hemorrhage.4 Type 2, or the paraprosthetic–enteric fistula does not develop a communication between the bowel and the graft and accounts for 15% to 20% of SAEFs.4 In this type of fistula, bleeding occurs from the edges of the eroded bowel by mechanical pulsations of the aortic graft. Sepsis is more frequently associated with this type of AEF (75% of cases).4 The mean time interval between surgery and presentation with SAEF is about 32 months6 but the time interval can vary from 2 days to 23 years.7 AEFs can involve any segment of the GI tract but, 75% involve the third part of the duodenum and the affected part is generally proximal to the aortic graft.8
The pathogenesis of AEF is not fully understood but two theories exist. One theory suggests repeated mechanical trauma between the pulsating aorta and duodenum causes fistula formation and the other suggests low-grade infection as the primary event with abscess formation and subsequent erosion through the bowel wall.9 The latter theory is felt to be most likely. The majority of grafts show signs of infection at the time of bleeding and up to 85% of cases have blood cultures positive for enteric organisms.10
The main symptom of AEF is GI bleeding. Secondary AEFs have been traditionally said to present with a symptom triad (as in our patient) of abdominal pain, GI bleeding and sepsis; however, only 30% of patients present in this manner.11 Patients often have a “herald bleed” which is defined as a brisk bleed associated with hypotension and hematemesis that stops spontaneously followed by massive gastro-intestinal haemorrhage in 20% – 100% of patients.8 Sometimes the GI bleeding can be intermittent.
The commonest investigations for diagnosis of AEFs are OGD, conventional contrast CT scan and angiography.12 OGD is often the initial investigation, as in any upper GI bleed mainly because of lack of clinical suspicion of the diagnosis. The endoscopic findings vary from those of a graft protruding through the bowel wall to fresh bleeding in distal duodenum to that of an adherent clot or extrinsic compression by a pulsating mass with a suture line protruding into the duodenum.13 Less than 40% of patients have signs of active bleeding at OGD.8 Conventional CT with contrast is widely available and most commonly performed to diagnose AEFs. Perigraft extravasation of contrast is a pathognomic sign of AEF and this may be associated with signs of graft infection i.e perigraft fluid and soft tissue thickening along with gas.12 Multi-detector CT and MRI are more sensitive diagnostic imaging tools with MRI now being used mainly in patients with renal failure to avoid the use of contrast.12
PAEFs can be treated with endovascular stent placement in selected cases especially in those who cannot tolerate emergency surgery.12 The treatment of choice in SAEFs is graft resection and establishment of an extra-anastomotic circulation with repair of the duodenal wall although overall survival rates vary from 30% to 70%.13
Conclusion
SAEFs are a catastrophic complication of aortic surgery. AEFs are relatively rare and need a high index of suspicion in the appropriate clinical situation in order to diagnose this condition. Left untreated they are universally fatal. Surgical repair carries a very high mortality.
A 55 year old white male with a history of hypertension, hyperlipidemia, smoking and transient ischaemic attacks was admitted to the hospital with worsening dyspneoa on exertion over a period of 6 weeks. He also reported significant weight loss, loss of appetite and fatigue over several weeks. Physical examination revealed tachycardia, and moderate respiratory distress with prominent jugular venous distention. Cardiac auscultation revealed normal S1 and loud P2. Also heard were an early diastolic heart sound ( tumour plop) and a mid-diastolic murmur at the apex. An ECG revealed evidence of left ventricular hypertrophy with repolarization abnormalities. A transthoracic echocardiogram (Figures 1 and 2) revealed a large, pedunculated, mobile left atrial mass measuring 3x4 cm, impinging on the mitral orifice with a mean gradient across the mitral valve of 15 mm Hg. Left ventricular systolic function was normal.
Figure 1: Parasternal long axis echocardiograph of the left atrial myxoma prolapsing into the mitral valve during diastole.
A diagnosis of probable left atrial myxoma was made. The patient had four episodes of syncope within 24 hrs, the first at 3: 53 am after returning from the bathroom, subsequently leading to cardiac arrest at 14:20 pm.
Figure 2: parasternal long axis echocardiograph showing the large left atrial myxoma during systole.
He was intubated and initiated on vasopressors. An emergent Left heart catheterization was performed prior to a referral for surgical excision, which revealed triple vessel coronary artery disease. During cardiac catheterization the patient became more hypotensive requiring an intra-aortic balloon pump. While arrangements were made for a referral for surgery, the patient’s clinical condition deteriorated rapidly and he went into pulseless electrical activity at 18:54 pm and could not be resuscitated. The patient’s death was presumably due to persistent intracardiac obstruction. On autopsy, the left atrial mass was identified as a haemorrhagic left atrial myxoma, 5x4x3.5cm in size attached by a stalk to an inter-atrial septum. Multiple organizsing thrombi were present in the 1tumour. Histology showed abundant ground substance with stellate myxoma cells and haemosiderin-laden macrophages (Figures 3 and 4). The cause of death was attributed to valvular “ball-valve” obstruction.
Figure 3: Histopathology of left atrial myxoma showing spindle shaped myxoma cells (white arrow) in a myxoid matrix (black arrow) and blood vessels (top arrow) (H & E 40X)
Figure 4: Histopathology of left atrial myxoma showing vascular spaces filled with relatively fresh blood and evidence of old bleeding (hemosiderin) suggesting repeated episodes of hemorrhage within the myxoma (H & E 4X)
Case 2
A 57 year old African American female presented with recurrent syncopal episodes and dyspnea on exertion, orthopnea, leg swelling, abdominal distention, loss of appetite and fatigue for the preceding nine months. Physical examination revealed jugular venous distention, a displaced apical cardiac impulse, a parasternal heave, and a loud S2. Also detected were a pan-systolic murmur at the lower left sternal border, an early diastolic heart sound with a mid diastolic murmur at the apex, bibasilar crackles, ascites, and oedema up to the thighs.
Significant laboratory values were a total bilirubin of 1.6 mg/dl, and B- Type Natriuretic Peptide of 1323 pg/ml. A chest x-ray revealed an enlarged cardiac silhouette, right lung atelectasis and effusion. An ECG revealed left atrial and right ventricular enlargement.
The patient was admitted with the diagnosis of new onset congestive heart failure and was treated with intravenous lasix, and fosinopril. A 2-D Echocardiogram revealed a large mass suggestive of myxoma in the left atrium measuring 4.5 x 7.5 cm, occupying the entire left atrium protruding through the mitral valve into the left ventricle (Figure 5) .
Figure 5: Apical four chamber echocardiograph of the left atrial myxoma prolapsing into the mitral valve during diastole.
This mass was obstructing flow with a mean trans mitral gradient of 17 mm Hg, with a reduced stroke volume and severe pulmonary hypertension with an estimated Right Ventricular systolic pressure of 120 mm hg. A presumptive diagnosis of left atrial myxoma was made and the patient was scheduled for its surgical removal the following morning. The patient was transferred to the intensive care unit for closer monitoring; and fosinopril and lasix were discontinued. At about 22:30 hours that night patient was noted to be hypotensive with systolic blood pressure of around 80mm Hg. The patient was treated with normal saline and concentrated albumin. She then developed acute respiratory distress at 23:00 hours requiring intubation and ventilator support. Intravenous dobutamine, dopamine and later norepinephrine were added for continued hypotension. The patient went into pulseless electrical activity, she was successfully coded with a return of her pulse but continued to be hypotensive. Cardiothoracic surgery decided not to take the patient for emergency surgery due to her unstable haemodynamic condition. The patient’s family was notified of the poor prognosis and the decision was made not to resuscitate her if her condition deteriorated further. The patient ultimately became bradycardic and went into asystole at 5: 30 am. An autopsy was not performed. The cause of death was attributed to large left atrial myxoma causing valvular obstruction and cardiovascular collapse.
Discussion
These two cases illustrate an uncommon, malignant course of a left atrial myxoma with rapid progression of symptoms which proved fatal. The most common primary tumour of the heart is myxoma accounting for 40-50% of primary cardiac tumours(2,3) .Nearly 90% of myxomas occur in the left atrium(3) .In over 50% of patients, left atrial myxoma causes symptoms of mitral stenosis or obstruction. Systemic embolic phenomena are known to occur in 30-40% of patients(3) .
Table 1. Summary of 17 published cases of sudden cardiac death associated with cardiac myxoma in adults (1950-2008)
Author/Reference
Year
No
Age
Gender
Symptoms
Interval Between Symptoms To Scd
Size Of Myxoma In Cm
Autopsy
Vassiliadis (8)
1997
1
17
M
Dizziness
3 months
6
yes
McAllister (10)
1978
5
40 to60
NA
NA
NA
5 to 6
yes
Cina (2)
1996
6
Below 40
NA
Embolic, syncope
16.6 months
5.7
yes
Puff (9)
1986
1
41
M
Syncope,
months
1.5
yes
Puff (9)
1986
1
19
F
Syncope
6 months
3
yes
Maruyama (7)
1999
1
20
M
Dizziness
1 day
8
None, Patient survived SCD; Myxoma resected
Turkman (6)
2007
1
73
M
DOE
months
8
yes
Ito (13)
1987
1
28
M
Syncope
7 days
NA
yes
NA: not available
Constitutional symptoms reported in approximately 20% of patients include myalgia, muscle weakness, athralgia, fever, fatigue, and weight loss. Around 20% of cardiac myxomas are asymptomatic (3) .Severe dizziness/syncope is experienced by approximately 20% of patients due to obstruction of the mitral valve. (4) Of all the symptoms associated with cardiac myxomas, syncope is one of the most ominous prognostic indicators.
Although sudden death is known to occur in patients with primary cardiac tumour it is rare and is estimated to constitute 0.01 to 0.005% of all sudden deaths (1). Association between sudden death and cardiac myxoma has been reported as early as 1953 by Madonia et al (5). A review of the literature on this subject between 1950 to2008 revealed 17 cases of sudden death attributed to cardiac myxoma in adults (1, 6, 7, 8, 9, 10, 13)(Table 1) .
In all patients with unexpected death syncope was a predominant presenting symptom and their age ranged from seventeen to seventy three. The majority of patients with sudden death were men even though the tumour is more common in women. The size of the tumour did not influence clinical presentation and in some reports of sudden cardiac death tumour was as small as 1.5 cm and without previous symptoms (3). Sudden death in myxoma is attributed to either severe acute disturbance in cardiac haemodynamics from cardiac obstruction (ball-valve syndrome) or to coronary embolization from the tumour. The latter is probably responsible for sudden death in patients with very small tumours. In the study of Alverez Sabin et al (11) the initial neurological manifestation was Transient Ischemic Attack (TIA), but in none of the patients’ was a diagnosis of myxoma made because of the initial neurological symptom. Even though cardiac myxomas are a rare cause of TIA and syncope, it is important to consider cardiac myxoma in the differential diagnosis of any patient with a TIA or syncope (11). The patients presented here had a TIA and recurrent syncope placing them at high risk for sudden death.
The timing of surgical excision of myxoma is not clear and it is not unusual for patients to die or experience a major complication while awaiting surgery (2, 12). Intraaortic balloon pump (IABP) use has been described in one case of left atrial myxoma and life-threatening cardiogenic shock with favorable outcome(14) .As illustrated by the cases presented here it is essential that surgery be performed urgently once it has been identified that a patient has a myxoma that is large enough to cause complete intracardiac obstruction.
Oesophageal compression by a vascular structure resulting in dysphagia is uncommon. There have been multiple reports in the medical literature of almost every major vascular structure in the chest causing some degree of oesophageal compression and subsequent dysphagia(1, 2). In 1794, David Bayford, described a case of a 62 year-old lady having dysphagia due to an aberrant right subclavian artery. He coined the term “dysphagia lusus naturae”, which means “Freak of nature” (3).
Pill-induced esophagitis is associated with the ingestion of many pills and presents an uncommon cause of erosive oesophagitis (4). Multiple different classes of drugs have been described to be hazardous to the oesophageal mucosa and cause pill-induced oesophagitis (5, 6). Although uncommon in itself, dysphagia lusoria has not been described in literature to present as pill-induced oesophagitis. We present the first case of dysphagia lusoria causing pill-induced oesophagi's by testosterone pills in a young healthy man. Pubmed review of the English medical literature has been conducted to discuss the epidemiology, pathogenesis and management of this uncommon disorder.
Case Presentation:
A 26 year-old man with no significant past medical history presented with 5 days of dysphagia (for both solids and liquids), odynophagia and retrosternal chest discomfort. He admitted to having occasional difficulty swallowing for past 2-3 months for solids only. He denied any heartburn, cough, regurgitation, loss of appetite, weight loss, fever, chills, haematemesis or melaena. He denied tobacco or alcohol use. 2 weeks prior to presentation he had started taking testosterone pills for body-building. Physical examination was completely unremarkable.
A barium oesophagram showed extrinsic oblique compression of the oesophagus at the level of the carina as it passes through the aorta. CT scan and MRI of the chest revealed a right-sided aortic arch crossing posteriorly to the oesophagus with proximal oesophageal dilatation consistent with Dysphagia Lusoria. Endoscopy noted erosive oesophagitis/distinct ulceration extending from 18cm into a pulsatile area of narrowing at 20 cm with normal mucosa visualized distally.
Biopsies revealed oesophageal squamous mucosa with marked acute inflammation, reactive changes and no evidence of viral inclusions. Surgical management was discussed with the patient, but given the short duration of symptoms and the patient's stable weight, providing symptomatic relief with lifestyle changes, together with a trial of medications such as proton pump inhibitors were considered. At 2 weeks follow-up whilst taking proton pump inhibitors and having discontinued the testosterone pills, the patient experienced complete resolution of symptoms.
Epidemiology:
Dysphagia Lusoria: Moltz et al, found that lusorian artery has a prevalence of about 0.7% in the general population, based on the post-mortem findings(7). Also, out of 1629 patients who underwent endoscopy for various reasons, 0.4% had a finding of a lusorian artery in a report from Fockens et al(8). It has also been concluded based on the autopsy results and retrospective analysis of patients’ symptoms during life that about 60-70% of these patients remain asymptomatic (7). Coughing, dysphagia, thoracic pain, syncope and Horner's syndrome may develop, but usually present in old-age(9).
Pill-Induced Oesophagitis: The data on pill-induced oesophagitis is rather limited. A Swedish study found an incidence of 4 cases per 100,000 population/year(10). Wright found the incidence of drug-induced oesophageal injury to be 3.9/100,000(11). This may be underestimated and does not include subclinical or misdiagnosed cases. Also, cases are reported selectively, due to clustering of cases, newly implicated pills or rare complications. The incidence today is probably much higher due to increased use of prescription medications, widespread use of endoscopy and an ageing population. All these factors limit our ability to correctly assess the true epidemiology of this iatrogenic disorder.
Pathogenesis
Dysphagia Lusoria: During embryological development, the aortic sac gives rise to six aortic arches and with further development the arterial pattern is modified and the fourth arch persists on both sides and some vessels regress. In right arch anomaly, the left arch atrophies and disappears whereas the right arch persists. If both arches persist, they form a double arch or a vascular ring encircling the trachea and oesophagus (12).
Pill-induced oesophagitis: Several lines of evidence confirm that oesophageal mucosal injury is caused by prolonged contact with the drug contents(13, 14) .On clinical grounds, patients frequently report a sensation of a pill stuck in the oesophagus before the development of symptoms and the frequent occurrence of symptoms after improper pill ingestion. Endoscopically, the evidence includes occasional observation of pill fragments at the site of injury, sharp demarcation of the injury site from the normal tissue and the frequent localization of the injury to the areas of oesophageal hypomotility or anatomic narrowing(4, 15). Therefore factors predisposing to the drug-induced oesophageal injury can be divided into two main categories: patient or oesophageal factors (16, 17, 18,19,20,21) and drug or pharmaceutical factors as shown in tables 1 and 2 (13,14,22,23,24).
Table 1: Patient/Esophageal facors for pill-induced esophagitis
Old Age
Decreased Salivation
Pill intake in recumbent position
Lack of adequate fluid intake with the drug
Structural abnormalities of esophagus
Hypomobility of the esophagus
Table 2: Drug related factors for pill-induced esophagitis
Chemical structure(sustained release pills, gelatinous surface)
Formal structure (capsule increases risk over the tablet)
Solubility
Simultaneous administration of multiple medications
It is important to note that most patients who experience pill-induced damage have no antecedent oesophageal disorder, neither obstructive nor neuromuscular (25). It is the combination of anatomic narrowing coupled with the caustic effects of the implicated drug that caused the oesophageal injury in our case. Although testosterone pills have never been reported to cause pill-induced oesophagitis, 6 cases of corticosteroid-induced oesophagitis have been described in the literature(26) .
Clinical Presentation
Dysphagia Lusoria: As previously mentioned the disorder remains asymptomatic in majority of the patients. Symptomatic adults usually present with dysphagia for solids, (91%), chest pain (20% or less). Less commonly, patients may have cough, thoracic pain or Horner’s syndrome (27,28). In infants, respiratory symptoms are the most predominant mode of clinical presentation. This is believed to be due to absence of tracheal rigidity, allowing for its compression with resulting stridor, wheezing, cyanosis etc (9) . Richter et al. reported average age of presentation to be 48 years (27). Various mechanisms to explain this delayed presentation have been proposed such as increased rigidity of the oesophagus, rigidity of the vessel wall due to atherosclerosis, aneurysm formation (especially Kommerell’s diverticulum), elongation of the aorta etc (9, 29,30) .
Pill-induced oesophagitis: Patients with pill-induced injury usually present with odynophagia, dysphagia and/or retrosternal chest pain(4) . Symptoms can occur after several days after starting a drug, but frequently occur after the first dose. (13) .Fever and haematemesis signifying a possible mediastinal extension can occur without chest pain(32,33). Pharyngitis due to the pill lodged in the hypopharynx has been reported(34).
Our case presents a typical example of asymptomatic Dysphagia Lusoria, who developed acute dysphagia, odynophagia and retrosternal chest discomfort immediately after the initiation of the offending agent; which is very typical of pill-induced oesophageal injury.
Diagnostic approach
Dysphagia Lusoria: The best method to diagnose an aberrant right subclavian artery presenting with difficulty swallowing is initially with a barium oesophagram followed by a CT or MRI scan. (27) .Angiography although considered gold standard for the diagnosis of vascular abnormalities is now largely supplanted by newer less invasive techniques such as CT or MR angiography. Upper endoscopy may reveal a pulsating compression of the posterior wall of the oesophagus as in our case(9, 27). Endoscopic ultrasound, especially with Doppler technology may be helpful to confirm the vascular nature of the abnormality(27). Oesophageal manometry usually shows non-specific findings. High peristaltic pressures have been reported in the proximal oesophagus above the level of the compression (9, 35).
Pill-induced oesophagitis: Barium studies can be normal, and slowing of barium column may be the only abnormality seen(31). Double contrast studies may however, increase the yield of a positive result(36). Kikendall et al. reported that endoscopy revealed the evidence of injury in all the patients (5) . Endoscopy most commonly reveals one or more discrete well demarcated ulcers with normal surrounding mucosa. Ulcers may range from pin-point to several centimetres in diameter(5). Biopsies, if performed, help to distinguish the condition from infection and neoplasia.
Our case shows a distinct oblique compression in the posterior wall of oesophagus on the barium study (fig 1) and classic findings on MR/CT with contrast which also excluded any other thoracic vascular abnormalities (fig 2-5). Endoscopic images of a shallow ulcer are shown in fig 6,7.
Figure 1- Barium esophagram depicting the extrinsic indentation of the esophagus as it crosses the aorta.
Figure 2- CT scan of the thorax demonstrating esophageal compression from a posteriorly placed aorta.
Figure 3- Magnetic resonance image showing esophageal compression with proximally dilated esophagus.
Figure 4- CT image showing the right sided origin of the aortic arch.
Figure 5- Three Dimensional image of the heart and the right sided approach of the arch.
Figure 6- Endoscopic view of esophageal inflammation at the site of compression.
Figure 7- Endoscopic image of the esophageal injury using narrow band imaging.
Treatment
Dysphagia Lusoria: The treatment of patients with DL primarily depends upon the severity of symptoms. Mild to moderate cases are managed by lifestyle and dietary changes such as eating slower, chewing well, sipping liquids, weight reduction and reassurance as in our case.(9,27) Janssen et al also reported in a series of 6 patients that 3/6 improved with proton-pump inhibitor alone or in combination with the prokinetic drug cisapride (9) .Severe symptoms and failure of medical therapy may need surgical evaluation and treatment. Richter et al. reported 14/24 patients who underwent surgical repair of the aberrant vessel for DL(27) . Bogliolo et al proposed endoscopic dilation as a temporary alternative to relieve symptoms in patients who are poor surgical candidates (37) .
Pill-induced oesophagitis: Most uncomplicated cases of pill-induced oesophagitis may heal spontaneously, with resolution of symptoms in a few days to a few weeks. Withdrawal of the offending drug and avoidance of topically irritating foods such as citrus fruits, alcohol is imperative to aid healing (4, 13). Sucralfate, topical anaesthetics, and acid suppression are often used to aid in relief of pain(4, 15). Rarely, in severe cases, parenteral nutrition or endoscopic dilation of chronic strictures may be required. (15, 25)
Conclusion
Our case demonstrates a typical case of DL presenting with pill-induced oesophagitis who responded to conservative and acid suppressive therapy. Identifying the risk factors and adequate patient education is the key to prevention.
A 75 year old woman with a history of prosthetic mitral valve replacement, atrial fibrillation & TIA on warfarin was scheduled for TURBT to be done under spinal anaesthetic. Warfarin was stopped one day prior to admission and heparin infusion commenced on admission, with target APTT 2.5 times the normal. Heparin was stopped 4 hours prior to the spinal anaesthetic, which was difficult due to ankylosing spondylitis and needed four attempts. However, after an atraumatic tap and good sensory motor block, surgery was commenced without incident. Post-operatively, the patient developed a lower respiratory tract infection for which co-amoxyclav was commenced. On the fourth day post-op, the patient developed sudden onset, right leg weakness and paraesthesia, with right lower limb power 3/5, decreased tone and absent reflexes, leading to the diagnosis of a spinal haematoma post spinal anaesthesia. However on further examination, she was also noted to be anaemic with a drop in haemoglobin to 6g/dl, with an INR of 3.4 and an acute renal impairment with a serum creatinine of 120. In addition, bruising in the right flank, abdominal pain and a right iliac fossa mass were also noted. An urgent MRI was booked, but as the patient was haemodynamically unstable, a CT scan was deemed more appropriate, which showed a retroperitoneal bleed into the right illio-psoas. This was confirmed with a spinal MRI done subsequently, which also ruled out any spinal haematoma. The patient was treated conservatively with 5units PCV and 3units FFP. Her clotting profile gradually normalised as did her renal function and her right sensory-motor deficit continues to improve.
Discussion:Retroperitoneal bleed The predilection for bleeding into the retroperitoneal space has not been fully explained but a unique weakness of the vascular and connective tissue has been suggested.2 It is also most commonly seen in association with patients on anticoagulation therapy or haemodialysis, or with bleeding abnormalities,3 and may represent one of the most serious and potentially lethal complications of anticoagulation therapy. The incidence of retroperitoneal haematoma has been reported at 0.6-6.6% of patients undergoing therapeutic anticoagulation.4, 5, 6 Warfarin, unfractionated and low-molecular weight heparin have all been implicated.7 The risk of bleeding during unfractionated heparin therapy has been estimated to be two- to five fold greater than that with warfarin.8 However, it is nonetheless important to note that the therapeutic index of warfarin is narrow 9 and anticoagulant control is easily deranged by drugs (such as antibiotics) and co-morbid factors such as renal or hepatic dysfunction. Frequent INR measurement is the best way to avoid haemorrhagic complications. Patients report lower abdominal or hip pain radiating to the groin or anterior thigh. Bleeding into the psoas muscle causes spasm and hip flexion and, as it extends, flank or thigh bruising may appear. Femoral nerve compression reduces quadriceps power and causes loss of knee jerk and paraesthesia in the area of cutaneous supply. CT scan is the investigation of choice10 but ultrasound is also sensitive and is more rapidly available. Delay in diagnosis is potentially fatal because severe haemorrhage can supervene. Locally the haematoma may cause ureteric obstruction and acute renal failure, or femoral nerve compression.11 (Both of which were seen in the case reported). Treatment options are surgery 12 and conservative management consisting of treating the anaemia associated with the bleed and correcting the coagulopathy.13 Options to treat the coagulopathy would mainly depend on how quickly correction is required, to what range and how long normal clotting indices would be safe in a patient on therapeutic or treatment anticoagulation. Fresh frozen plasma (FFP at a dose 15ml/kg) is given for rapid but short-lived correction with the usual risks of transfusion of blood products. Vitamin K (>2.5mg) is given for a slower but more prolonged correction (leaving patients with artificial valves at risk of thromboembolic events and valve failure). Over-anticoagulation due to warfarin can be reversed completely and immediately by infusion of a complex concentrate of factors 2, 7, 9 and 10.14Spinal haematoma The true incidence of spinal haematoma is unknown and due to its rarity it is very difficultto evaluate risk factors prospectively and any properly poweredstudy would require many thousands of patients to investigatethis. Therefore, data on the incidence of spinal haematoma followingneuraxial blockade are mainly based on audit studies and casereports. Tryba15 reported that the incidence of spinal haematoma afterepidural and spinal anaesthesia is 1 in 150,000 and 1 in 220,000, respectively. The insertion and removal of an epidural catheter appeared to be of far greater importance in the genesis of a spinal hematoma.16, 17 The incidence of spontaneous spinal haematomais rarer still and is estimated at 1 patient per 1,000,000 patientsper year. 18 Central neuraxial blockade has a low incidence of major complications, many of which resolve within 6 months. 19 The symptoms of an acute spinal hematoma include a sharp irradiating back pain of radicular character, and sensory and motor deficits which outlast the expected duration of the anaesthetic. Not all of these symptoms have to be present at the same time. The clinical suspicion can only be confirmed by means of an emergency CT-scan (with myelography) or magnetic resonance imaging.20 The only treatment of a compressing spinal hematoma is an emergency decompressive laminectomy with evacuation of the hematoma. Final neurologic outcome depends on21, 22 the speed with which the hematoma develops; the severity of the preoperative neurologic deficit; the size of the hematoma; and most importantly, the time span between hematoma formation and surgical decompression. Complete recovery of neurologic function is possible if surgery is performed within 8 hours of the onset of the paraplegia. Conclusion The aim of this report is in no way to undermine the importance of Alderman’s advice to suspect the spine as an area of bleeding in patients on anticoagulant therapy. The above case is a reminder to consider retroperitoneal bleeding as one of the differential diagnoses of spinal haematoma in an anticoagulated patient who develops sudden onset spinal pain, with or without neurological deficit post spinal anaesthetic. The presenting symptoms are similar and early management is equally important in terms of associated morbidity when management is delayed.
The herniation of a gravid uterus through an incisional hernia site is a rare occurrence. Incisional hernia is a frequent complication of abdominal wall closure and the management of pregnancy with a large incisional hernia with gravid uterus in its sac is challenging. The following is a case report of gravid uterus through an incisional hernia of a midline incision. Case Report Mrs LB, 35 years, Parity 2, period of amenorrhea of 34 weeks 3 days, married for 12 years was admitted to the hospital from the outpatient department due to the ulceration of abdominal skin as a result of herniation of gravid uterus through the midline longitudinal incision of a previous caesarean section . She was a booked case of our hospital and had been receivingantenatal care since 20 weeks of gestation. At 20 weeks there was no herniation of the uterus through the incision line. In her subsequent visits she came with the uterus protruding through the incisional hernia. She was referred to the General Surgeon who recommended elective Caesarean section with repair of hernia. Her past obstetric history revealed that she had her first emergency caesarean section eight years before because of a breech presentation and a second caesarean section, due to thepremature rupture of membranes at term. Both the babies were living & well. On both occasions she was operated on through infra umbilical midline vertical incision. There was no history of caesarean section wound infection during the post operative period in the previous two pregnancies. On examination, she was moderately built and adequately nourished. There was mild pallor. Her pulse rate was 88 beats per minute and her blood pressure was 126/86 mm Hg. Heart and chest were normal. Abdominal examination revealed distention of the abdomen in thecentral area. The uterus was felt just underneath the skin with acomplete lack of anterior abdominal wall. (Figure I) Figure 1- Photograph showing gravid uterus lying in the incisional hernia sac The overlying skin was necrosed with evidence of ulceration and the presence of engorged veins. The fetus was lying in the herniated gravid uterus outside the abdominal cavity. Routine investigations were within normal limits. Ultrasound examination showed the uterus herniated in the incisional hernia of the anterior abdominal wall with the live fetus in cephalic presentation without any gross congenital malformation. The placenta was located in the upper uterine segment. She was kept in the hospital for bed rest with abdominal support. Emollients & antiseptic skin ointment were applied over the skin of the anterior abdominal wall. An elective caesarean section was planned for 37 weeks but she went into labour at 36 weeks. The abdomen was opened by elliptical incision. The uterus was visualized just beneath the skin and there was no evidence of the rectus sheath in the vicinity of the incision. A uterineincision was made over the previous caesarean scar and the baby was delivered with APGAR 7/10 at 1 minute and 9/10 at 5 minutes. The uterus was repaired in layers and a bilateral tubal ligation was done. Herniorraphy was performed in double buttress fashion. She was given a course of antibiotics. Her post operative period was uneventful and she went home with a healthy baby weighing 2.25 Kg. During her follow up visits she was found to be problem free. Discussion The remote complication of a caesarean section could be an incisional hernia due to defective abdominal wound healing and herniation of gravid uterus through the abdominal wall. This is a rare complication.1 The complications that have been reported in literature in association with this complication include strangulation, abortion, pre-term labour, accidental haemorrhage, intrauterine fetal death and rupture of the lower uterine segment.2 Excessive stretching of the skin may cause ulceration of the skin as in this present case due to friction between the hernia sac and other parts of the patient’s body. Caesarean section should be performed and herniorrhaphy can be performed during the caesarean section as in the present case.1 Herniorrhaphy can be performed during pregnancy if there is evidence of morbid incarceration or the skin is necrosed.3 However, herniorrhaphy can be postponed until delivery, as the enlarged uterus may interfere with healing of the repair.
Bronchogenic cysts are lesions of congenital origin derived from the primitive foregut. They form due to ectopic budding of the foregut during the first trimester. Epithelial cells of the developing trachea and lung are pinched off and grow separately from the airways. Bronchogenic cysts are most commonly mediastinal, unilocular and contain clear fluid. Clinically, most cysts are symptomatic and occur in infancy or early childhood. Respiratory distress is the most common presentation in paediatric patients, manifested by recurring episodes of cough, stridor, and wheezing. Patient Description: A 13-year-old female presented with a two month history of right-sided back pain and five days of intermittent fever. The pain was worse on inspiration and made sleeping difficult. She denied wheezing, chest pain, or cough. She continued daily participation in competitive sports. Previous trials of antibiotics and an inhaled bronchodilator for presumed exercise-induced asthma were unsuccessful. Chest x-ray (CXR) showed a large cyst (10x10x8 cm3) in the posterior right lung. (Image 1). Image 1: Initial chest x-ray revealing bronchogenic cyst in the posterior right middle lobe (10x10x8 cm3) Computerized tomography (CT) scan showed a large cystic lesion arising entirely within the right lower lobe and extending the width of the hemithorax. (Image 2) Image 2: Chest CT shows bronchogenic cyst extending the entire width of the right hemithorax and approximately 50% full of fluid. There was an air-fluid level occupying ~50% of the cavity. She was diagnosed with a multilocular bronchogenic cyst. She was briefly hospitalized and discharged on azithromycin with plans to resect the cyst in one month. Severe cough, fever, and chills prompted readmission after 3 weeks of antibiotic therapy. CXR and CT showed cyst enlargement (16x9x11 cm3) with over 95% fluid. (Images 3 and 4) Image 3: Substantial bronchogenic cyst (16x9x11 cm3), over 95% full of fluid. Image 4: Lateral chest x-ray revealed opacification along superior margin of cyst. She was started on ampicillin/sulbactam. Percutaneous drain placement yielded a large volume of turbid fluid. Aerobe, anaerobe and fungal studies of the fluid were negative. Resection was postponed due to significant inflammation surrounding the cyst cavity. She was discharged on a seven day course of amoxicillin/clavulanate. Following six weeks of cyst drainage, a thoracoscopic right lower lobectomy was performed. Extensive inflammation and induration made dissection of the lower lobe and pulmonary vessels challenging. Fibrinoid adhesions extended to the pleural surface. Operative time was 418 minutes. Surgical pathology showed diffuse necrotizing granulomatous inflammation with acid-fast bacilli and multiple nodules up to 3.3 cm in diameter. Ninety-five percent of the pleural surface had nodular involvement. (Image 5) Areas of non-indurated lung also showed small nodules with a miliary appearance. Inflammation was present at the bronchovascular margins, hilar nodes, and distal lung. Image 5: Gross specimen of right lower lobe: Approximately half of the lobe was indurated and 95% of surfaces showed nodular involvement. Sectioning through indurated region revealed diffuse nodules up to 3.3 cm. Nonindurated lung showed small nodules with miliary appearance. The patient had no history of tuberculosis exposure, foreign travel or immunodeficiency. There was no family history of tuberculosis or respiratory disease. Based on the acid-fast bacilli identified on pathology stain, fluid drained from her chest tube was sent for acid-fast bacilli culture and smear. Mycobacterium was not isolated. It was determined that the source of the atypical mycobacterial infection was likely colonizing mycobacteria from her oropharynx that became entrapped in the cyst. A six-week course of clarithromycin, rifampin, and ethambutol was prescribed to treat any remaining organisms. At two-month follow-up, she had minimal pulmonary symptoms and inflammatory markers were improved. Erythrocyte sedimentation rate (normal: 0-15) and C-reactive protein level (normal: 0-10) decreased from 88 and 173 during her hospitalization, to 10 and 3.6, respectively. At four-month follow-up, she had resumed competitive sports and had no evidence of ongoing infection. Discussion: This case highlights a unique presentation of infected bronchogenic cyst after substantial cyst growth. Unusual aspects include the late onset of symptoms, multilocular intraparenchymal cyst appearance, turbid drainage, extensive nodularity, necrotizing granulomatous inflammation, and atypical Mycobacterium infection. Although comorbid infection is not uncommon, causative organisms are typically Haemophilus influenzae1,2 and Streptococcus pneumoniae.3 Cases of Streptococcus pyogenes,4 Escherichia coli,5and Salmonella enteritidis6have been reported. However, only four cases of bronchogenic cyst with Mycobacterium infection have been documented.7,8,9 Three of the Mycobacterium-infected cases are adult patients. Lin et. al reported a 39-year-old female with bronchogenic cyst complicated by Mycobacterium avium infection.7 The organism was identified by genetic sequencing of biopsied lung tissue. Sputum acid-fast stain and mycobacterial cultures were negative. Liman et al. reported two adult cases: a 20 year-old male with Mycobacterium identified in a right lower lobe specimen but with negative sputum culture, and a 32 year-old female with Mycobacterium isolated in a sputum culture but a negative microscopic exam and cyst fluid culture.8 The only documented paediatric case, a 9 year-old female with a 6 cm right lower lobe bronchogenic cyst, was reported by Houser et al.9 She underwent lobectomy; Kinyoun stain of the cyst specimen showed Mycobacterium. Sputum culture and acid-fast bacilli stain were negative. Tuberculin skin test was positive. Comorbid infection with Mycobacterium tuberculosis was suggested, but they were unable to isolate an organism. Treatment consisted of four months of rifampin and two years of isoniazid with pyridoxine. This is the first documented paediatric case of bronchogenic cyst infected with atypical Mycobacterium. Her presentation is noteworthy, given the substantially greater size of the cyst (16x9x11 cm), extensive pathologic findings, and success with a different antibiotic regimen.
Bronchogenic cyst should be included in the differential diagnosis of a child with cough, dyspnoea, and fever. Although rare, we stress the importance of keeping mycobacterial infection in mind in cases of an infected cyst. Acid-fast culture should be done on sputum and cyst contents. Due to the frequency of negative cultures, stains should also be performed on resected cyst specimens. Antibiotic therapy should be considered and administered based on the extent of infection. All symptomatic or enlarging cysts warrant surgical excision. Prophylactic removal of asymptomatic cysts is recommended due to higher rates of perioperative complications once cysts become symptomatic.10 We raise the question of whether earlier CXR is indicated to rule out bronchogenic cyst, particularly when patients do not improve after trials of watchful waiting, antibiotics, and bronchodilators for other possible respiratory diagnoses.




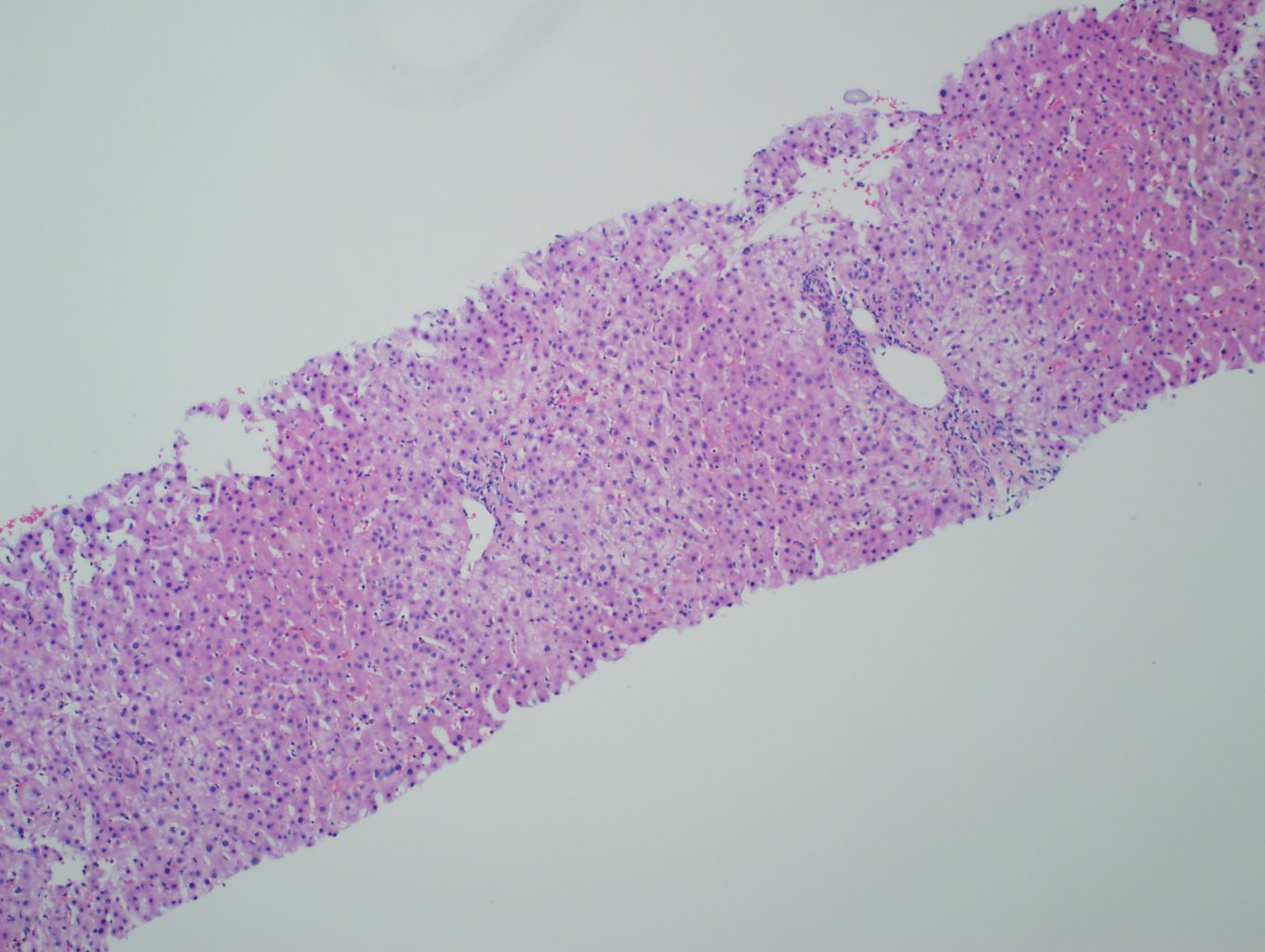
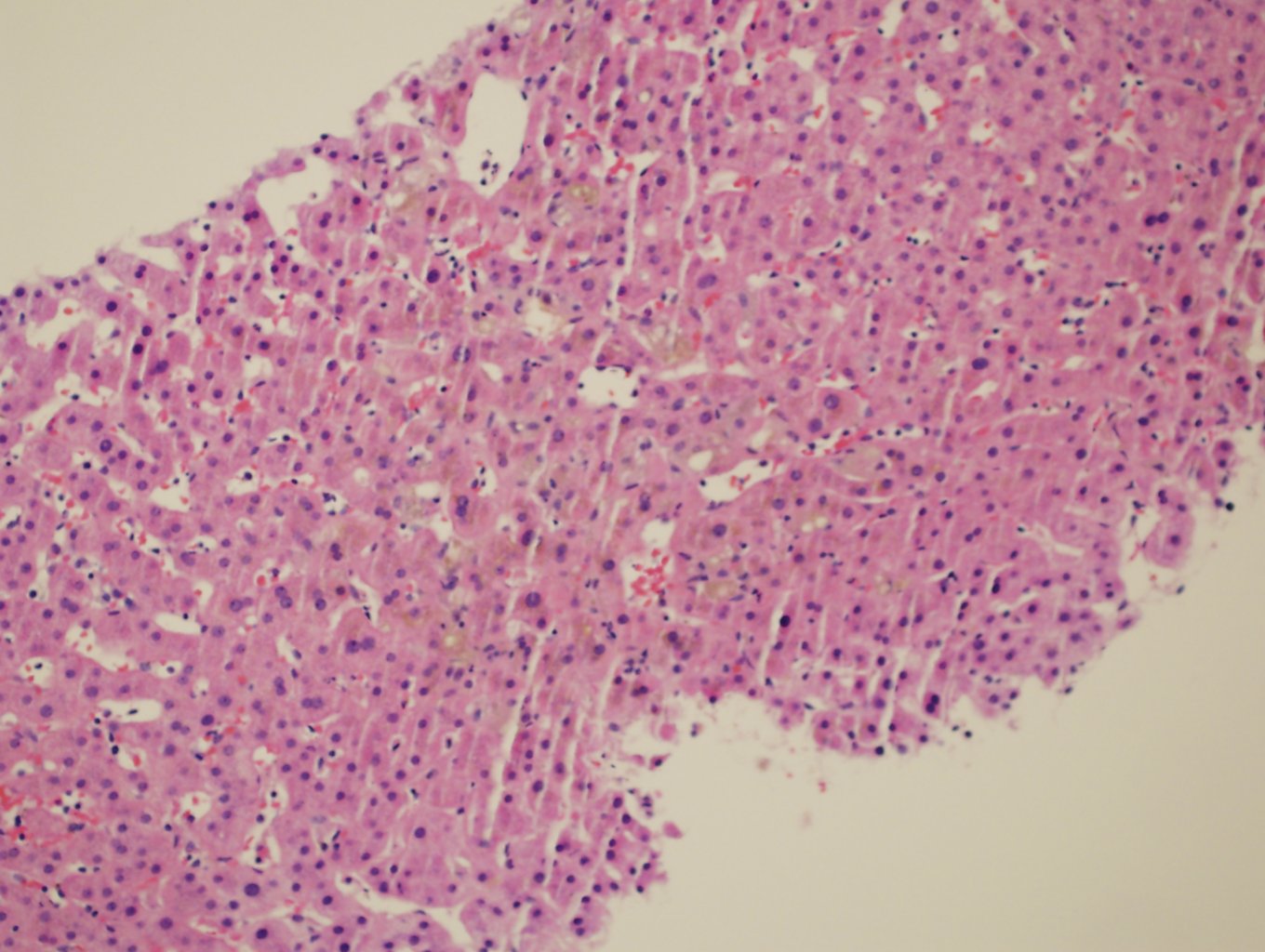
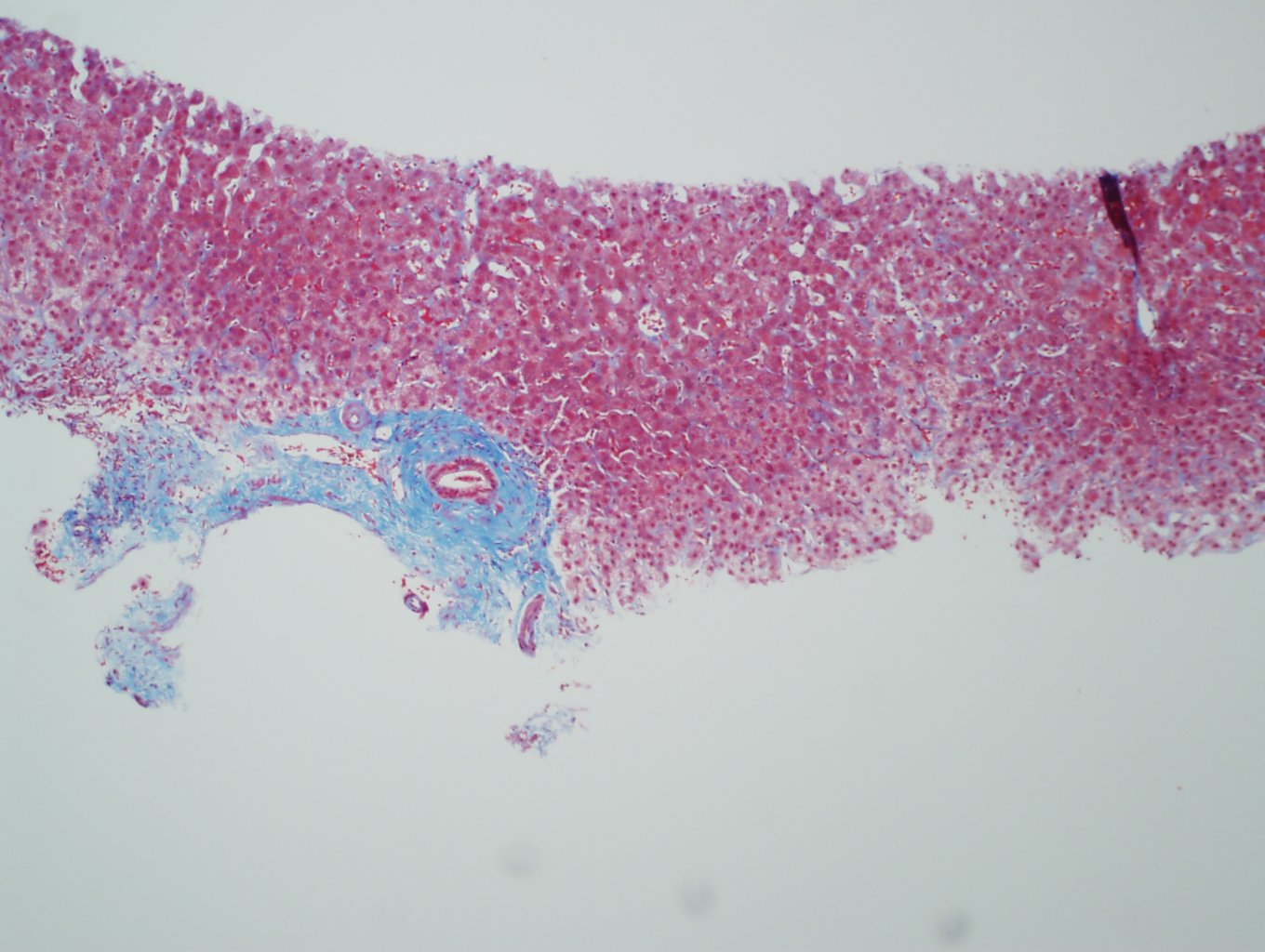
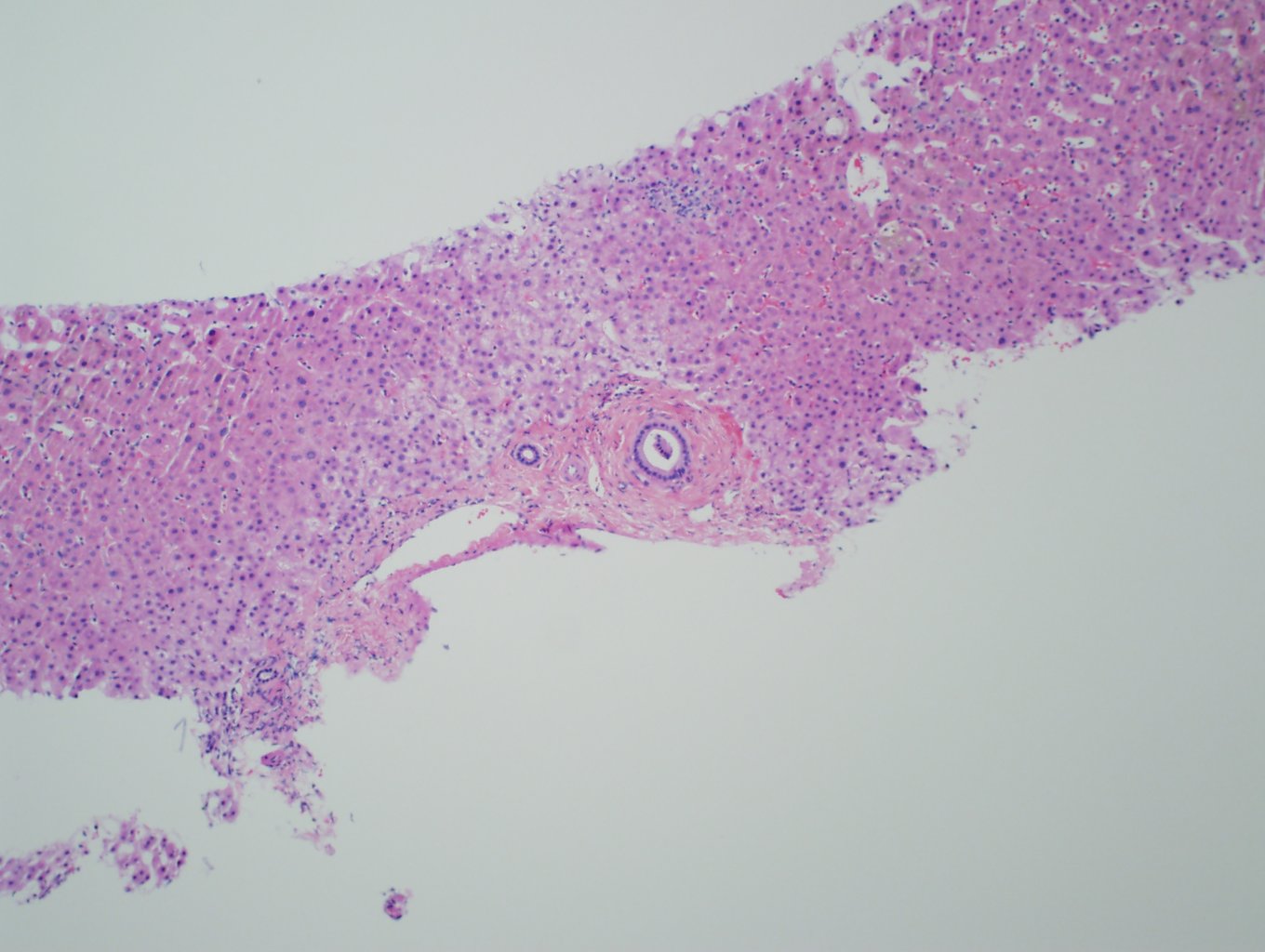
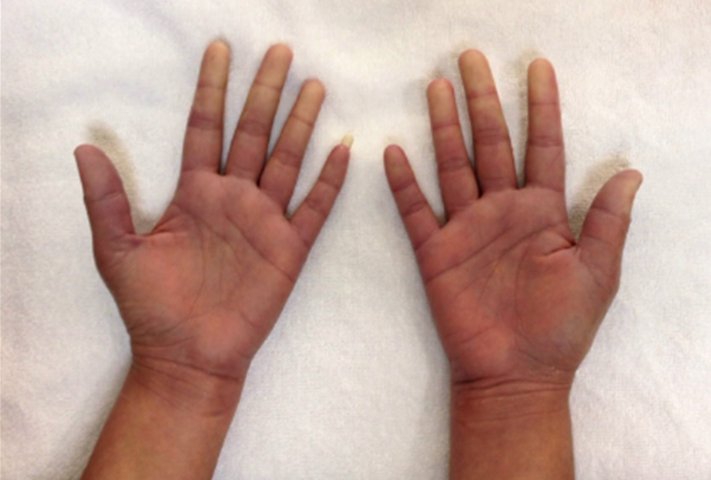
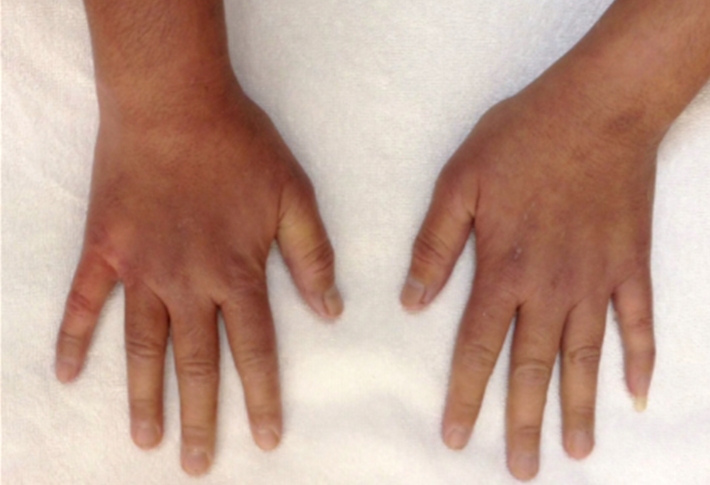
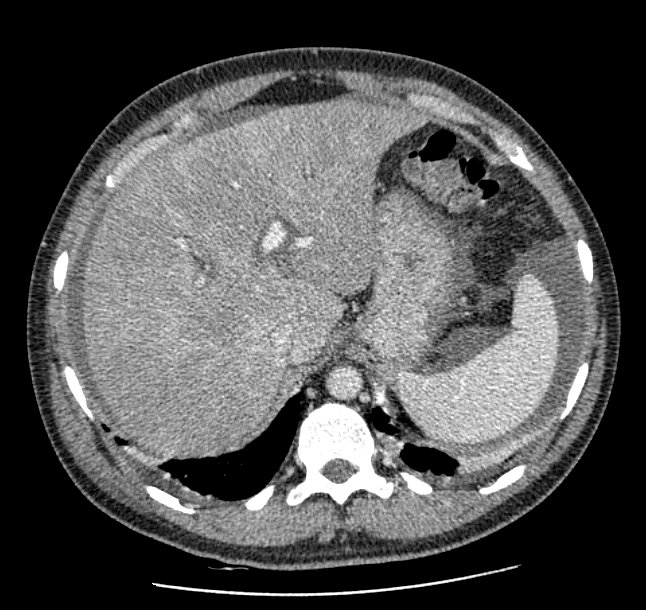
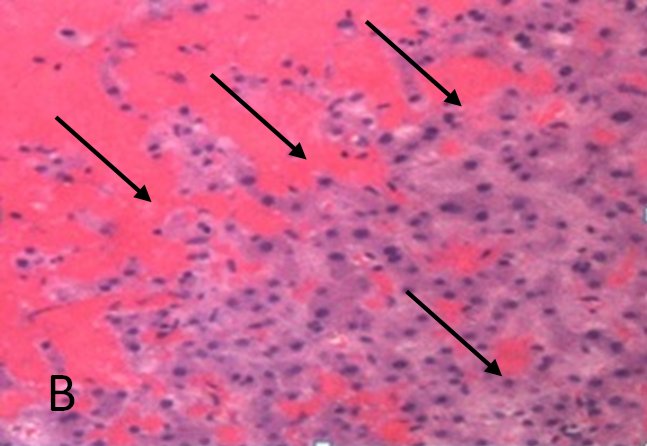
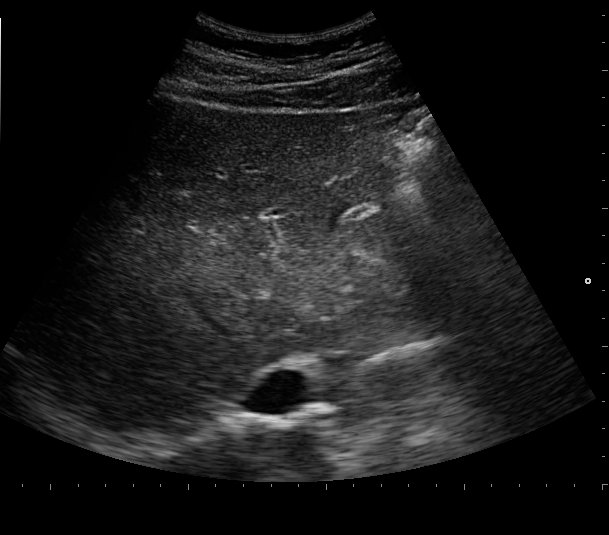
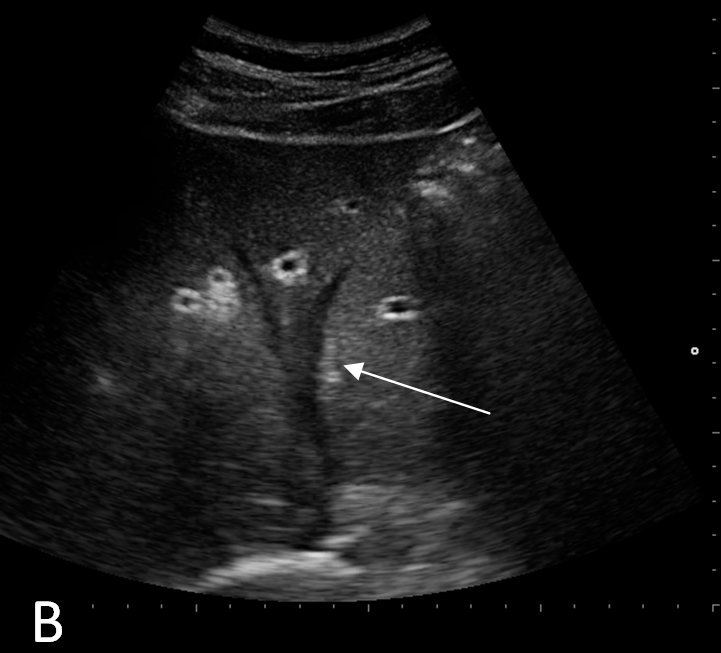
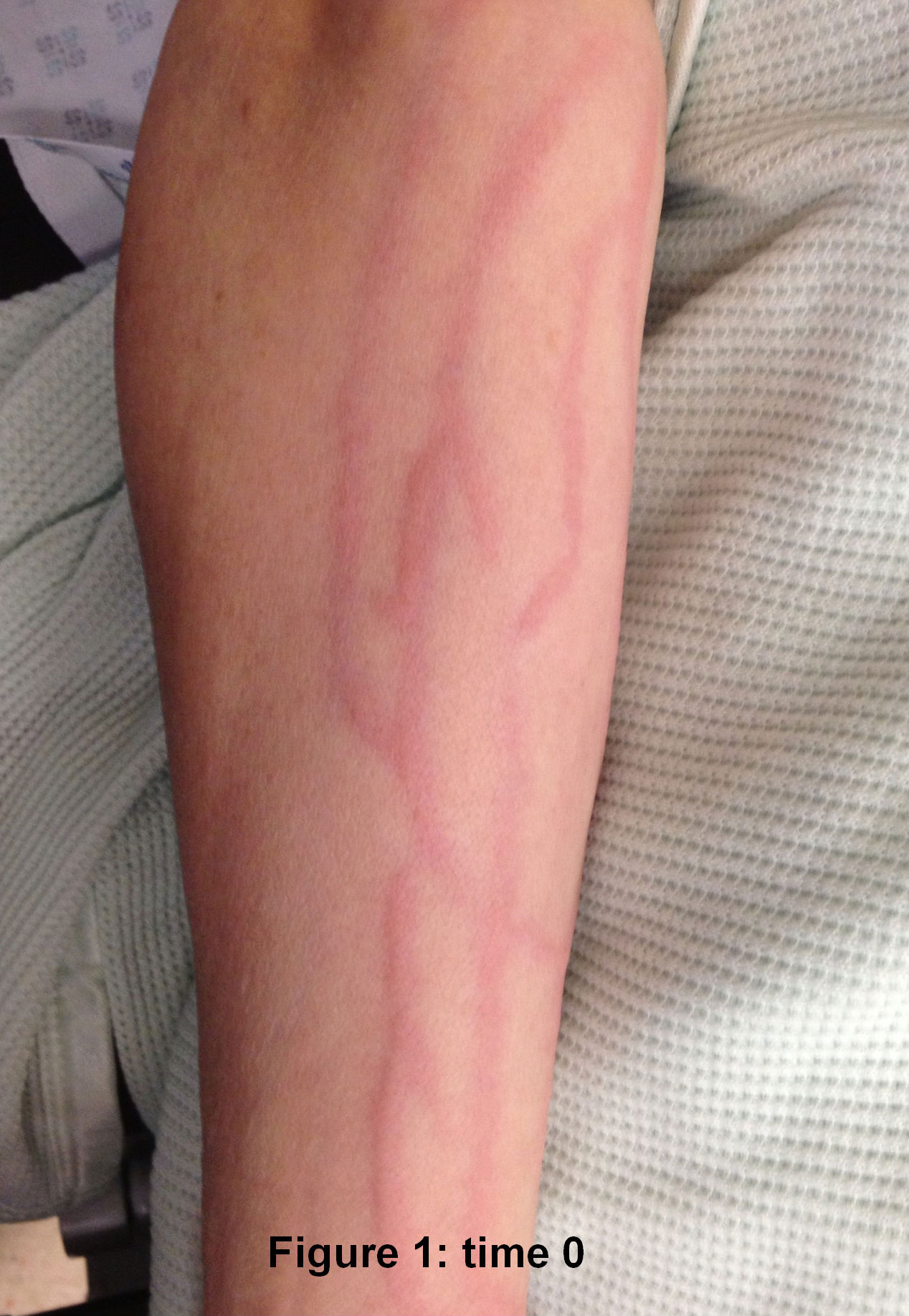
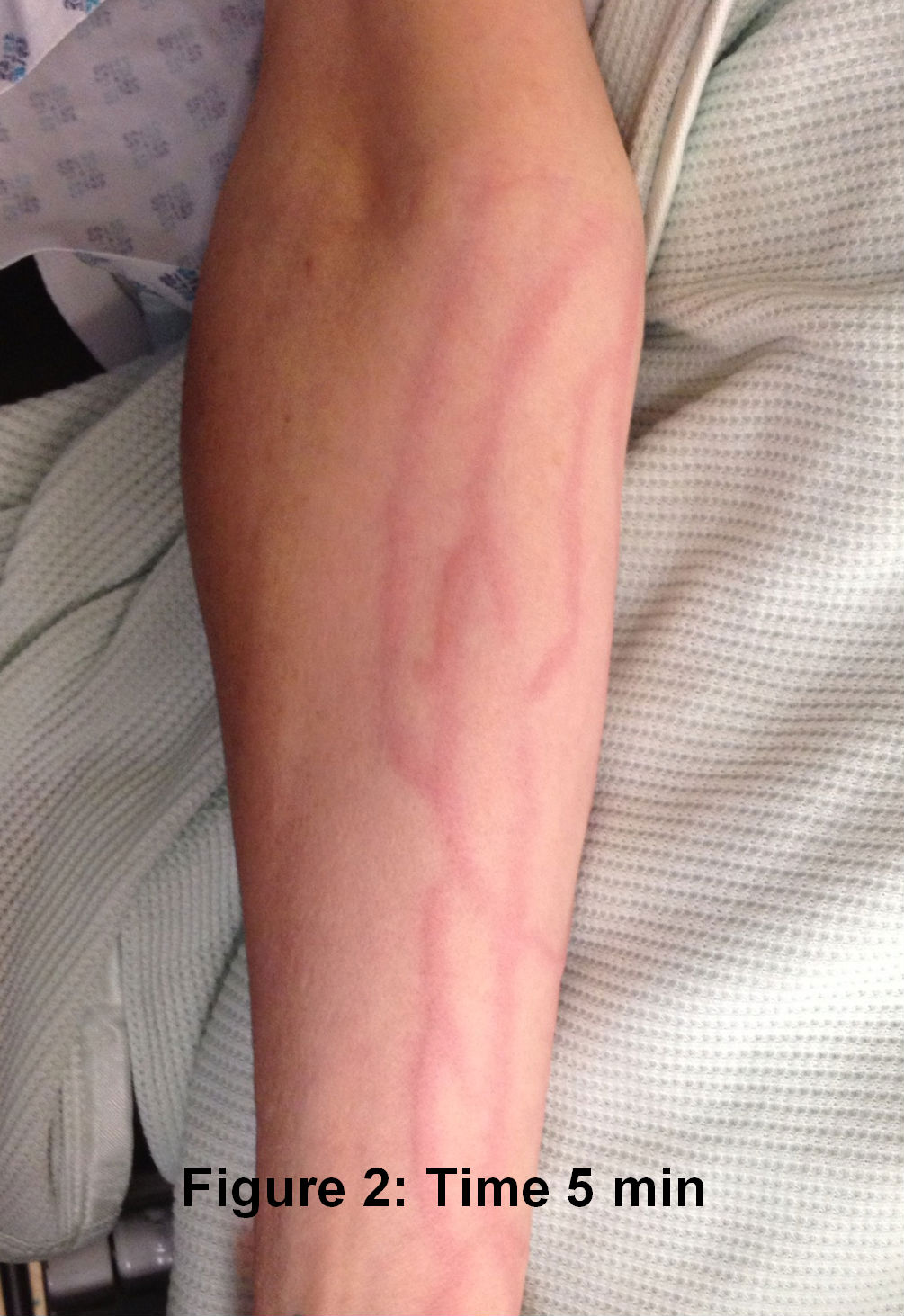
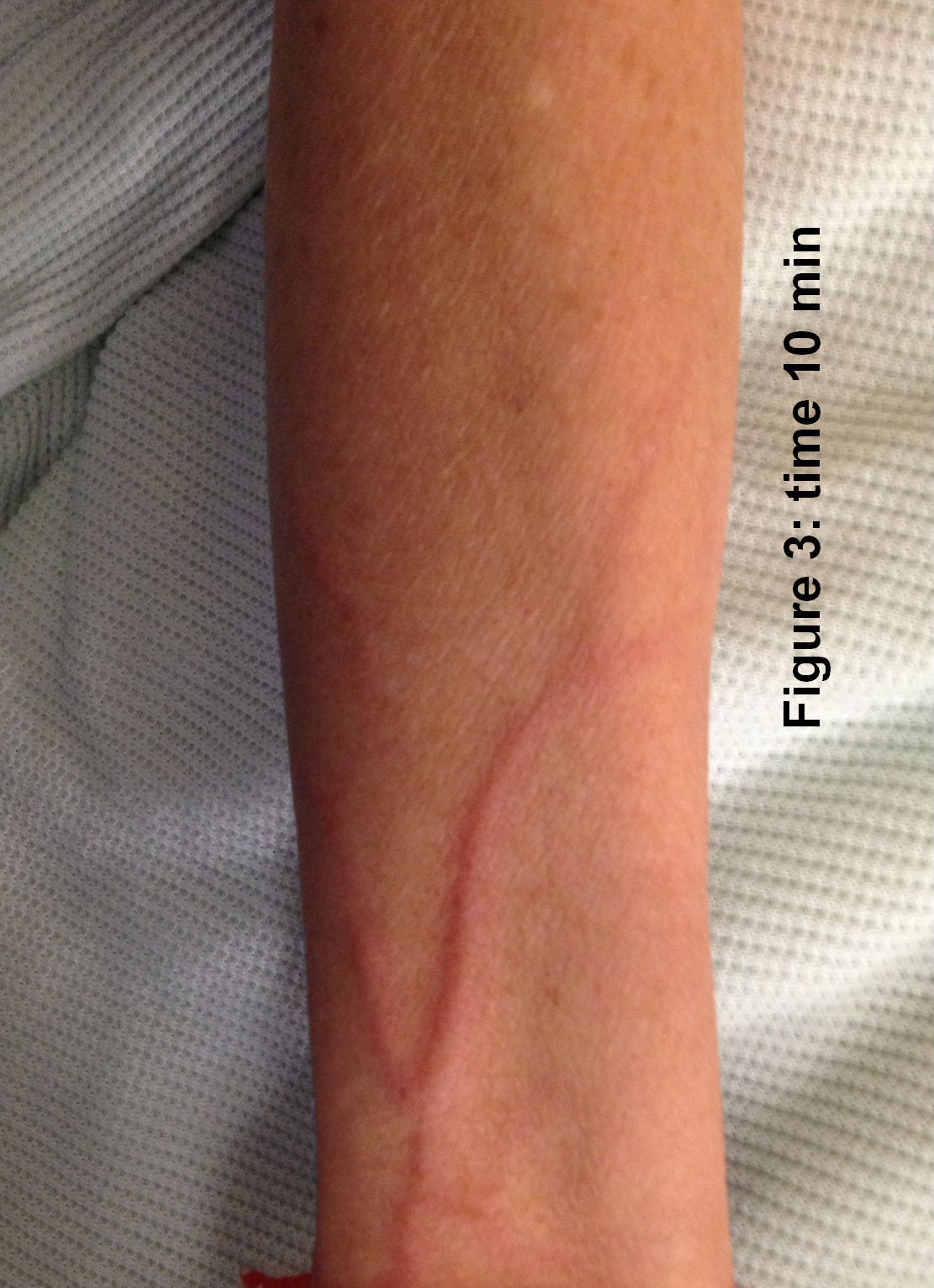
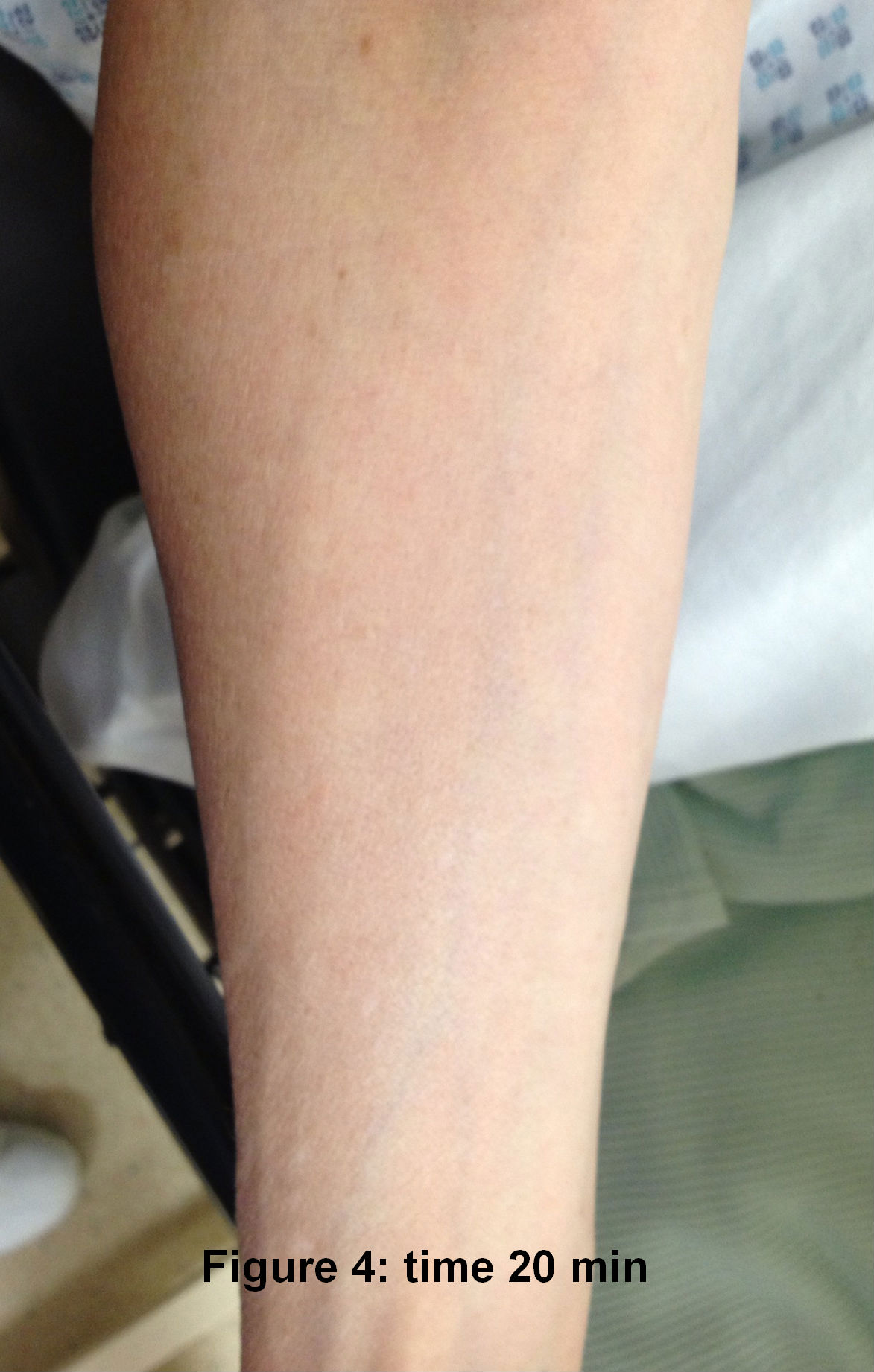
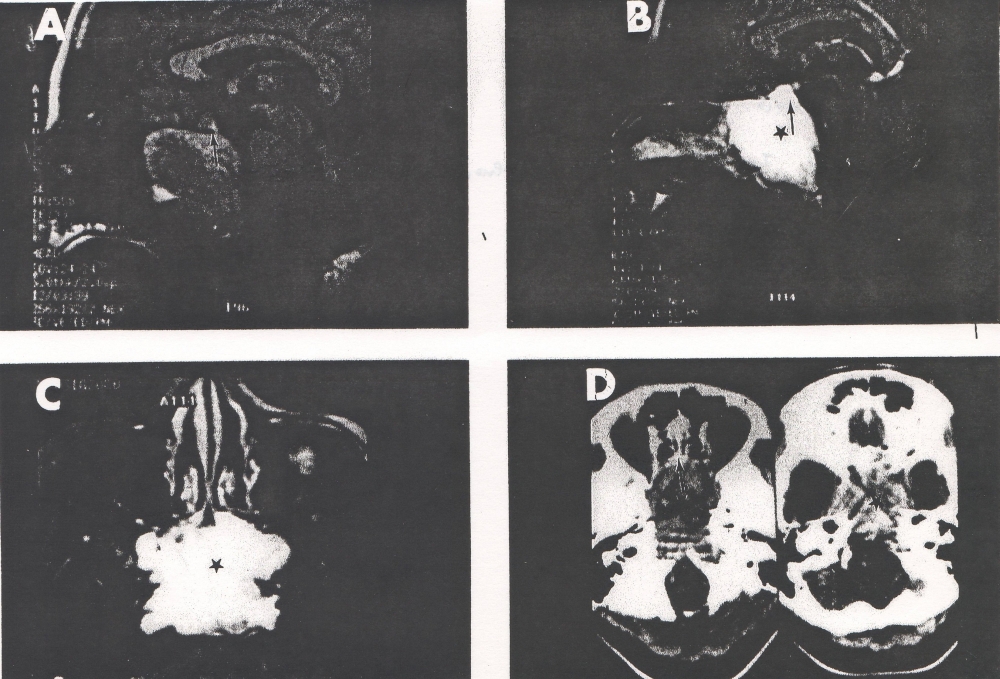
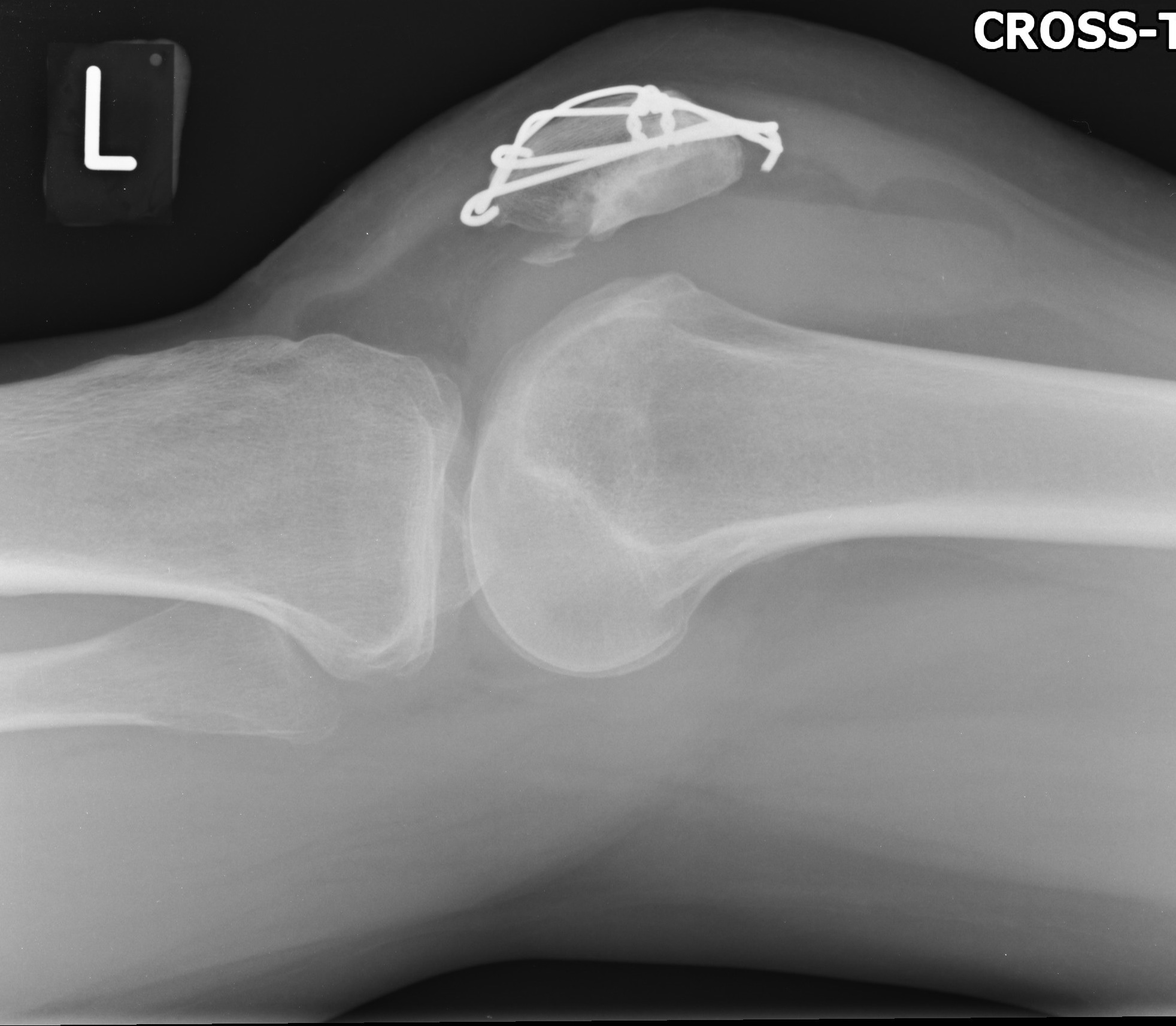
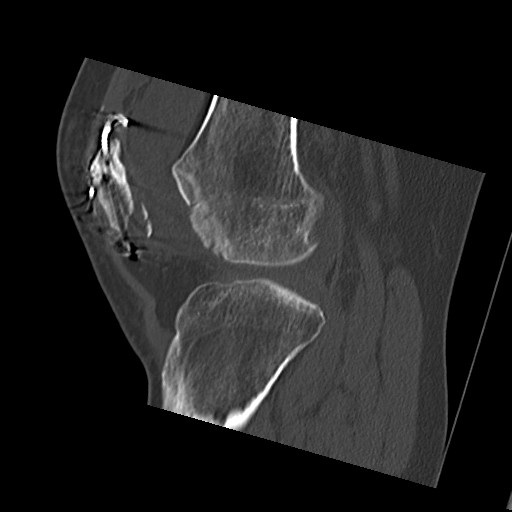
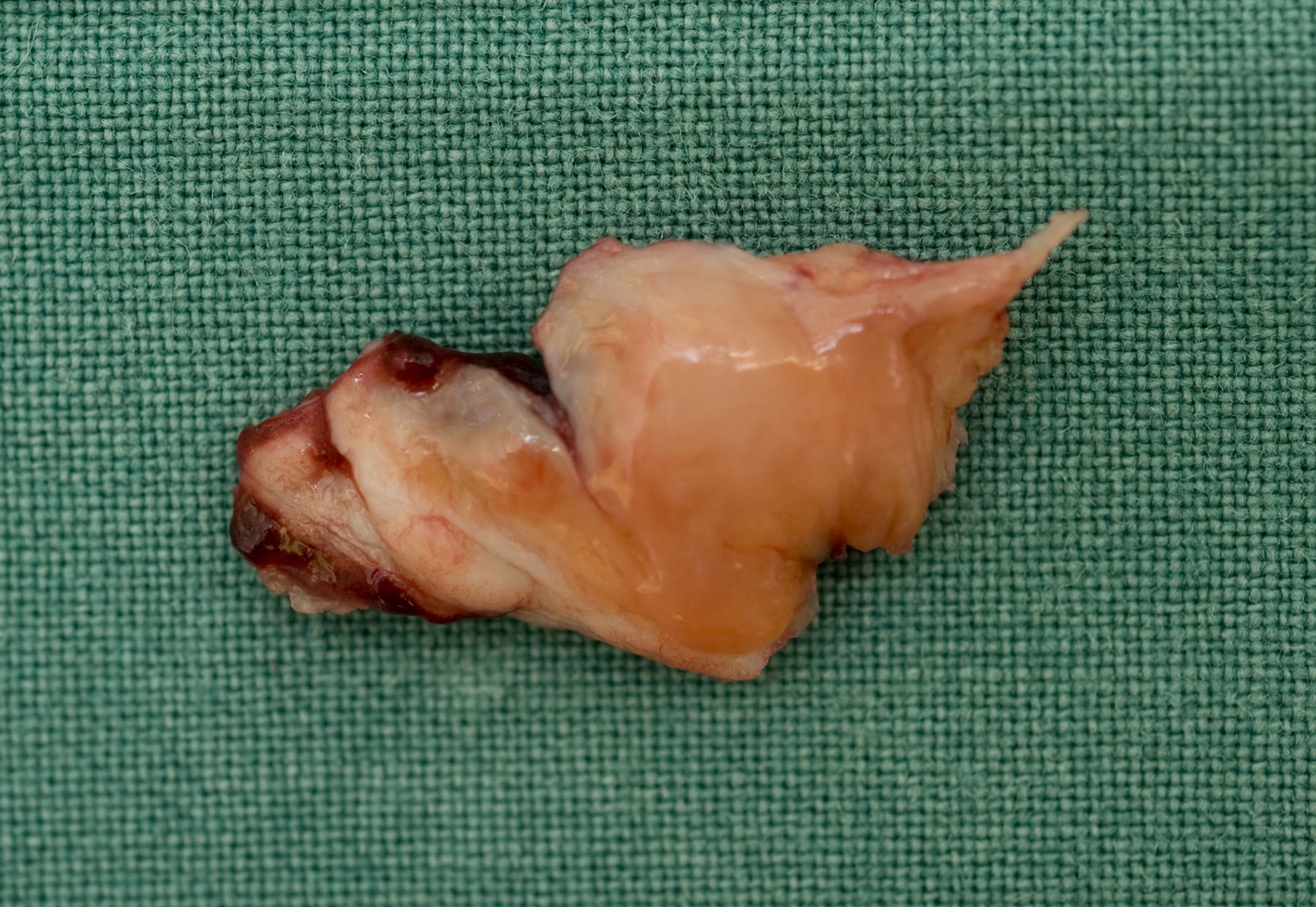
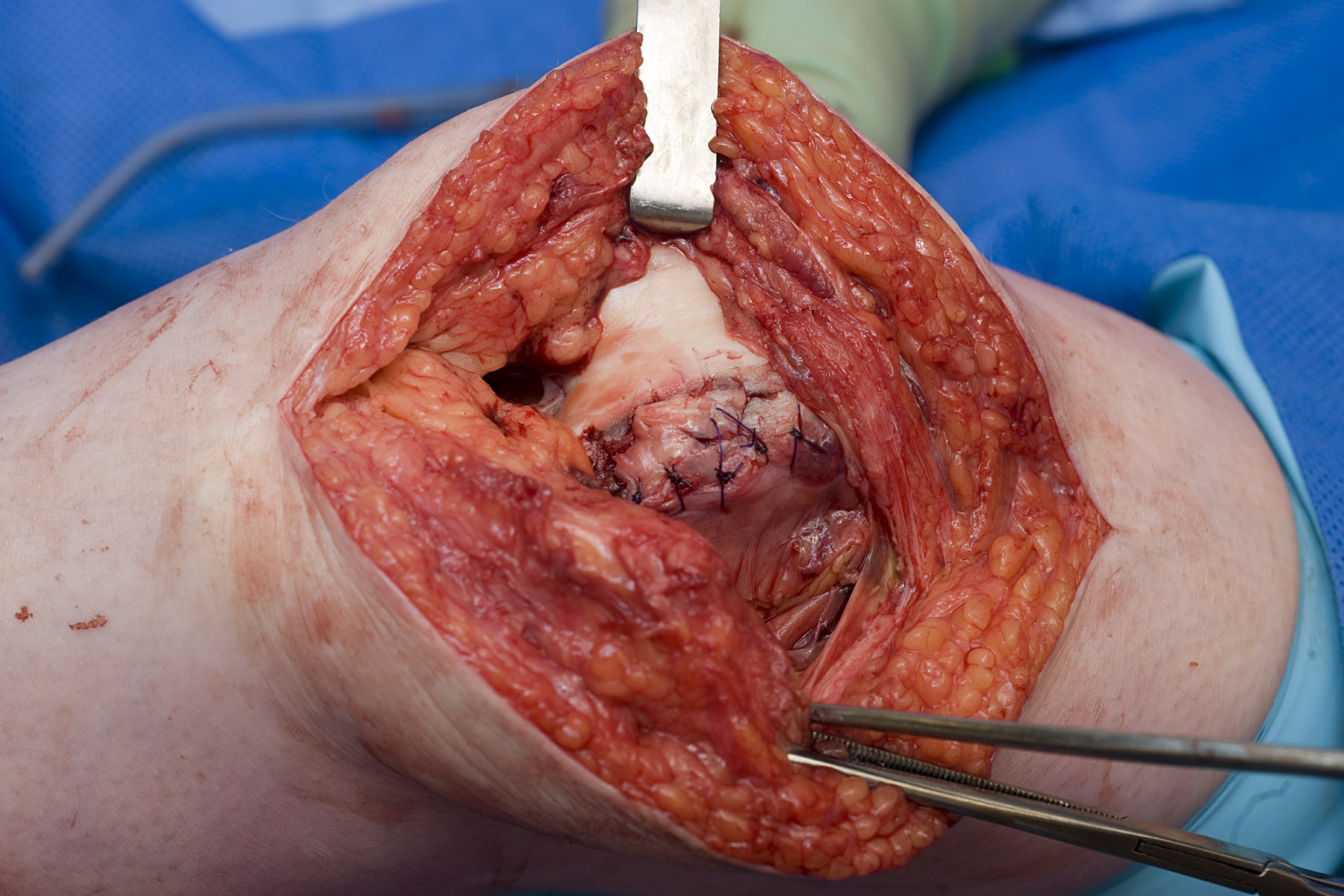
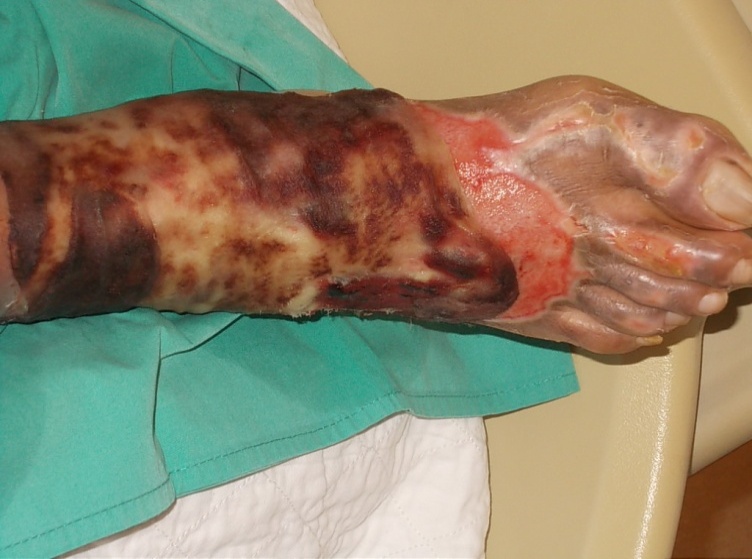
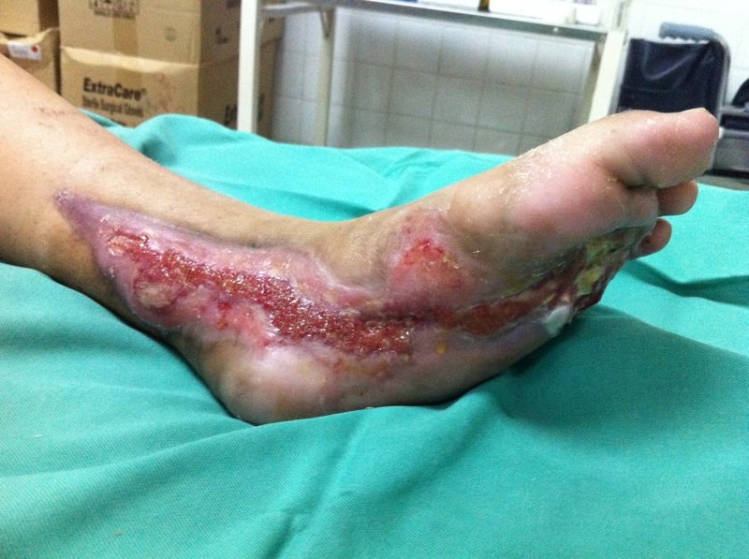
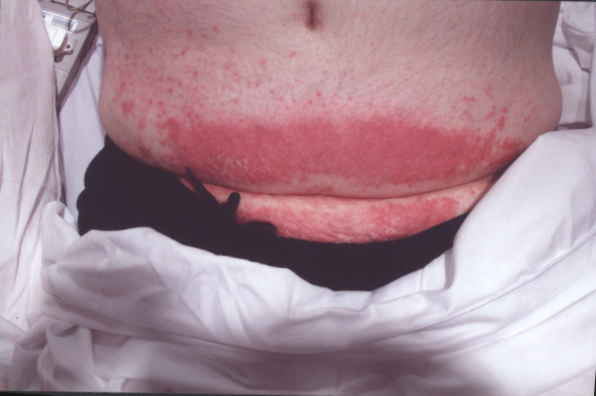
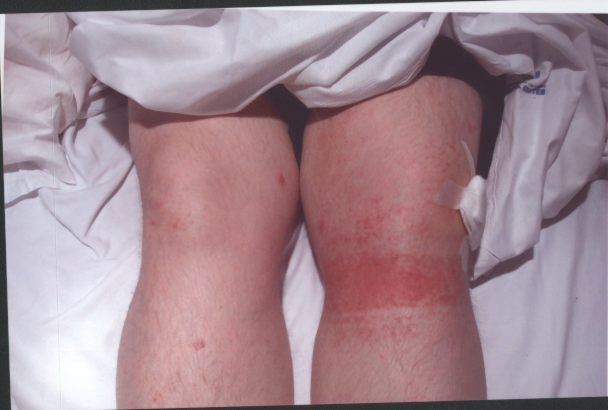
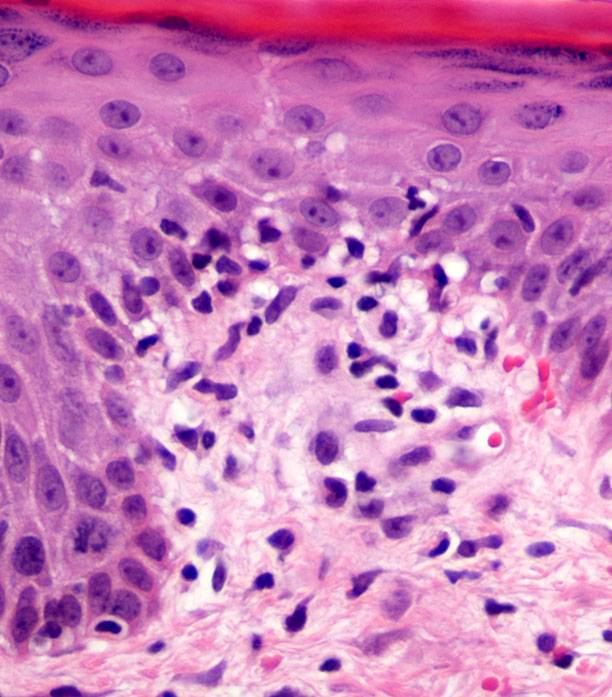
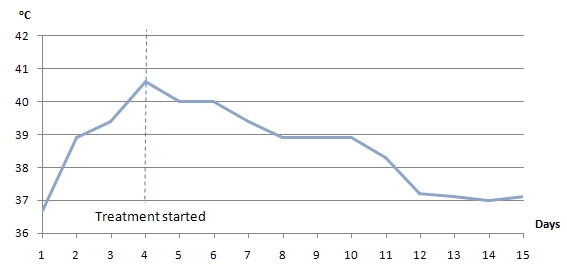
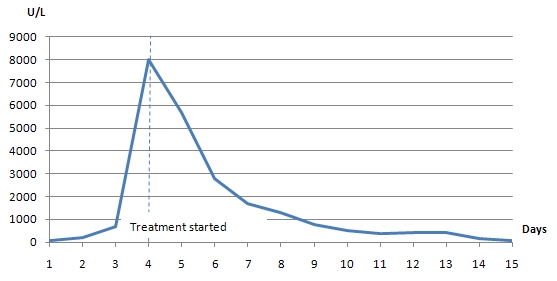
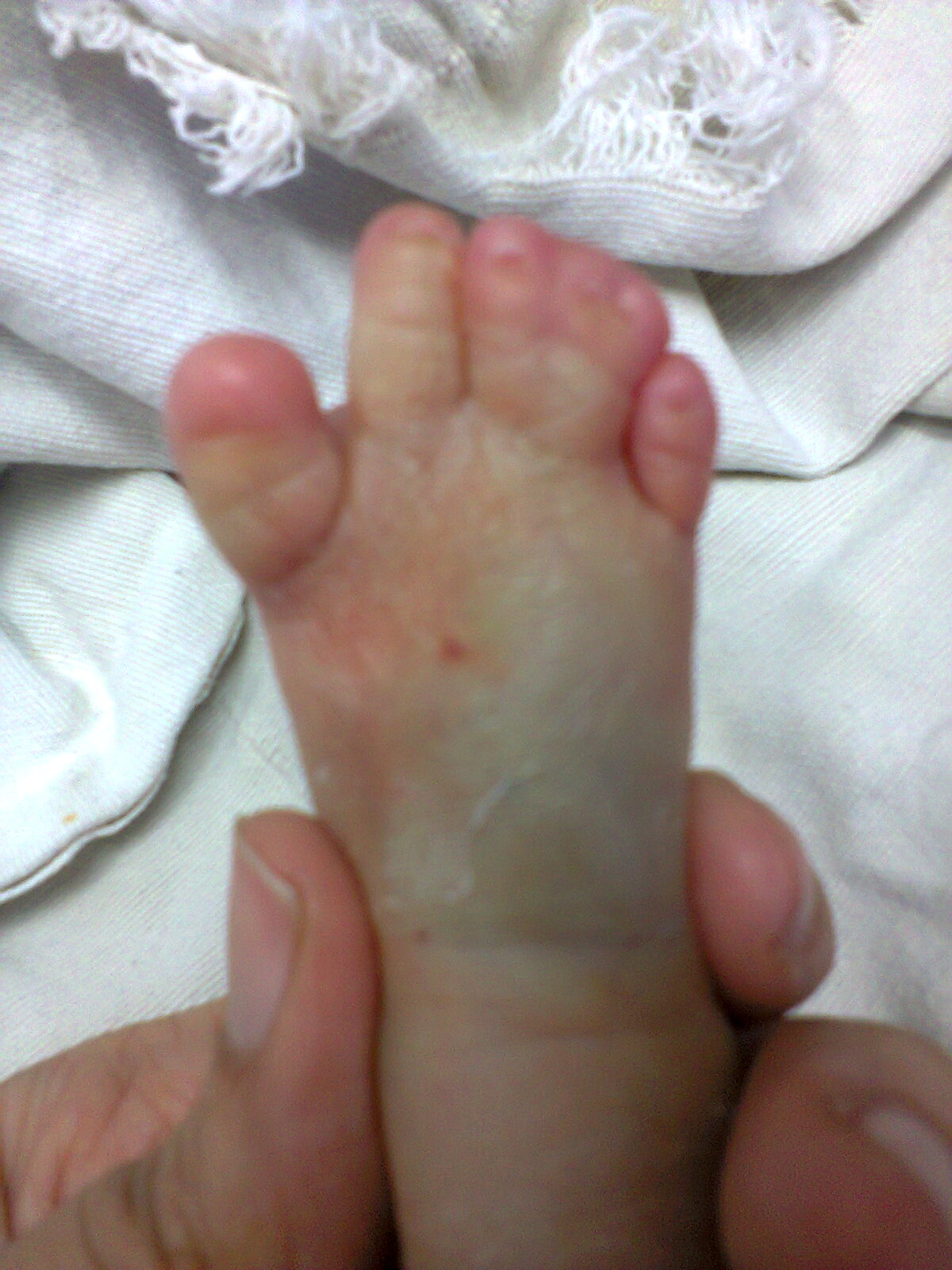
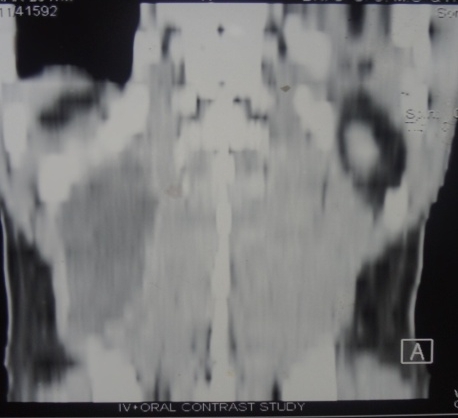
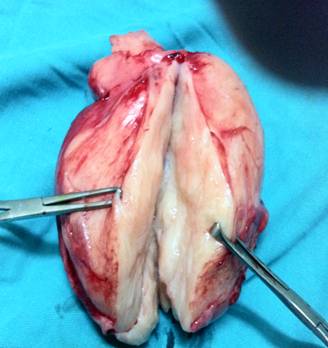
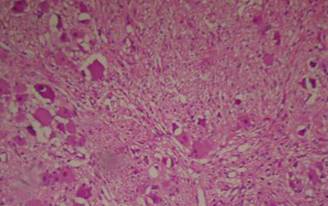
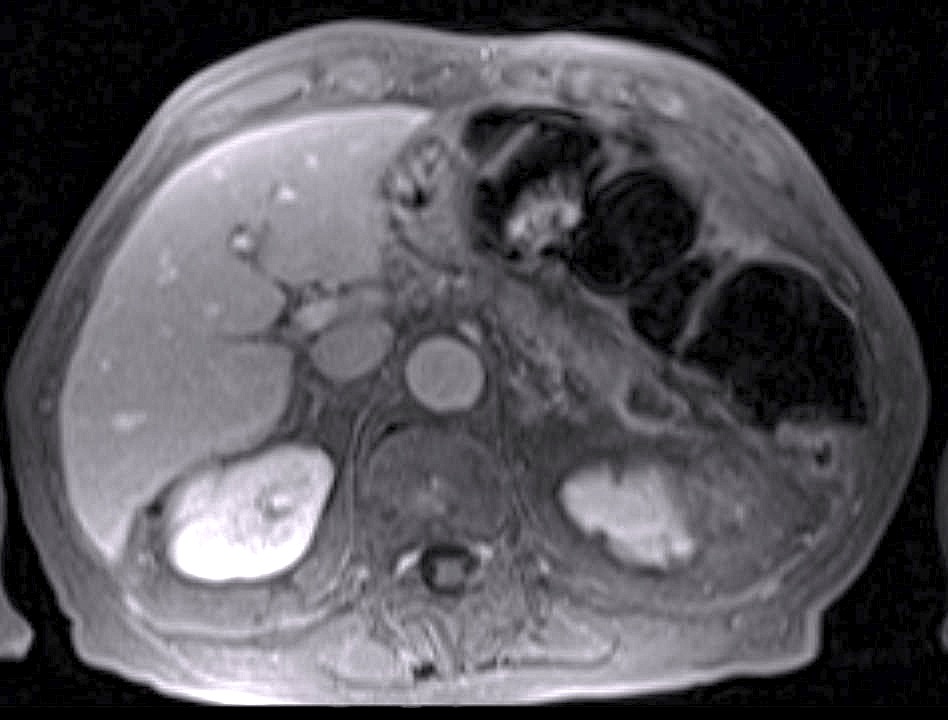
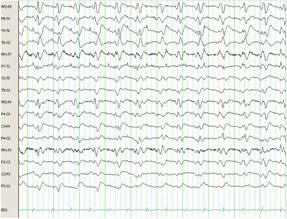
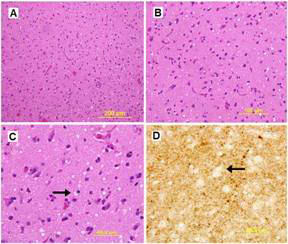
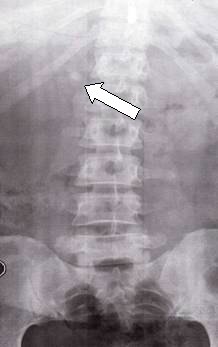
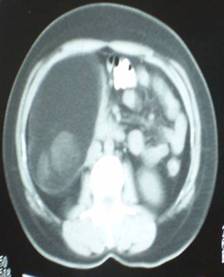
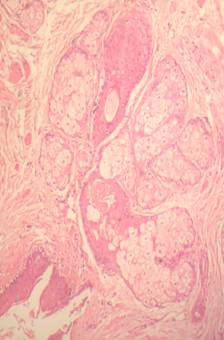
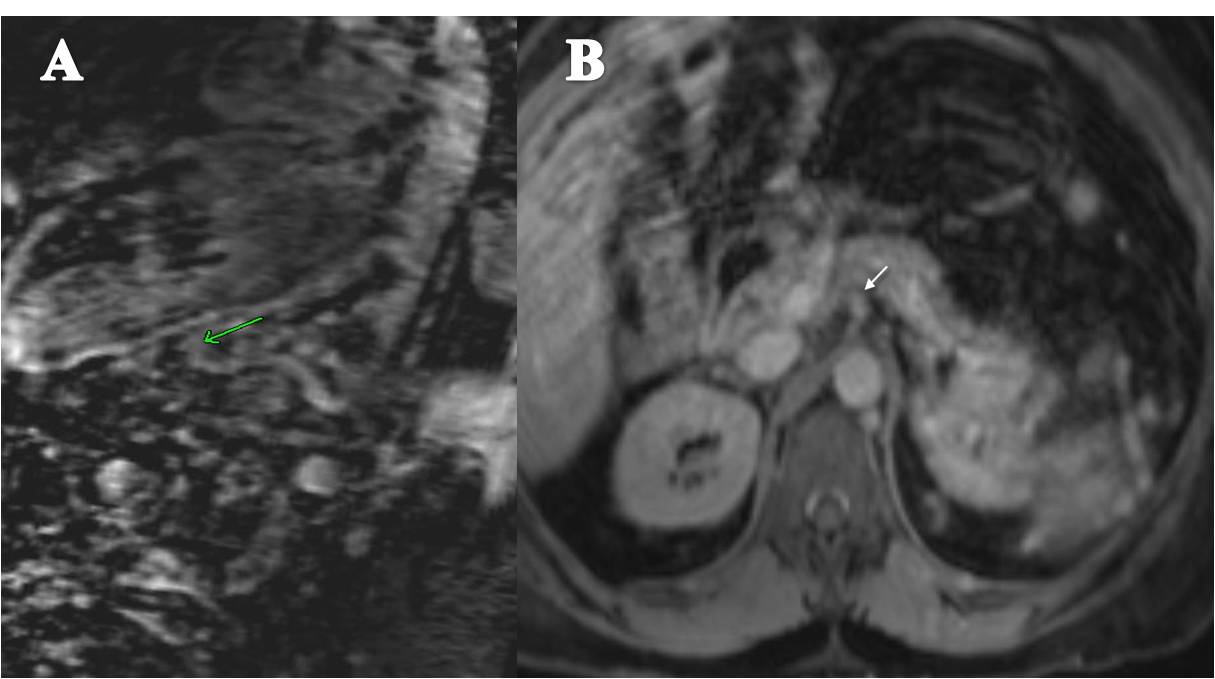
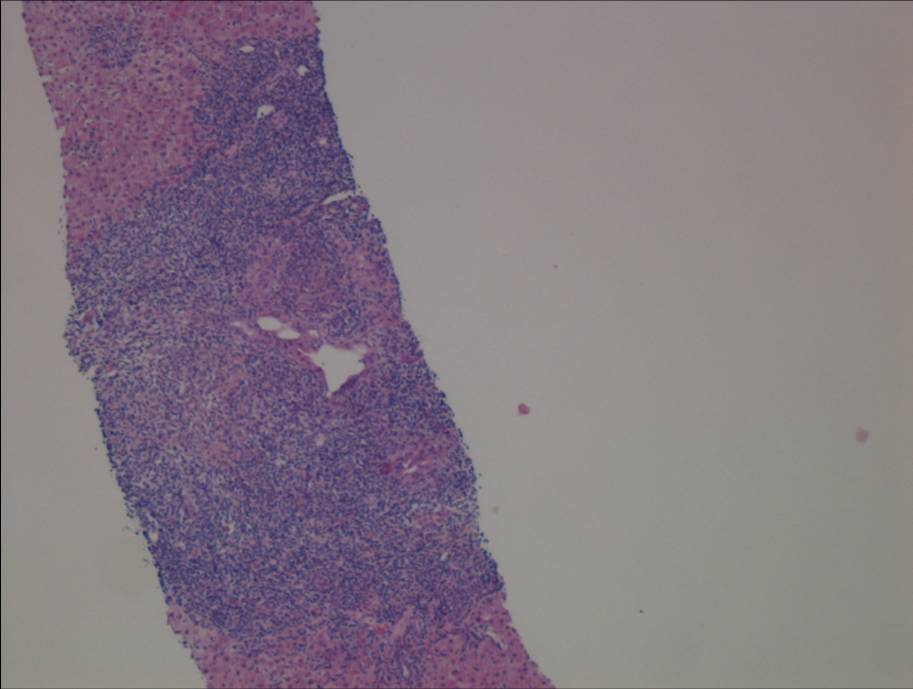
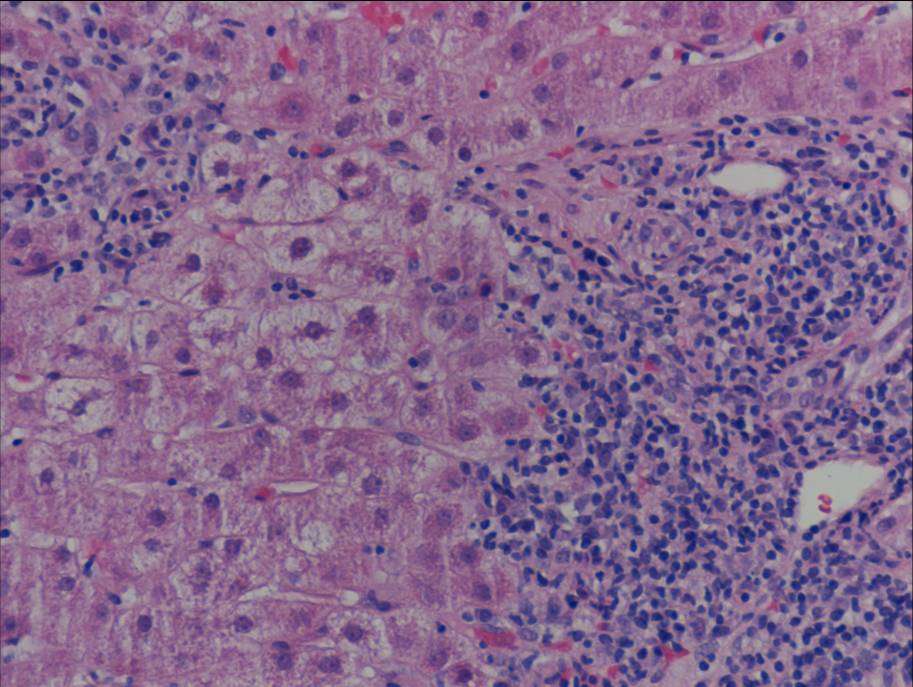
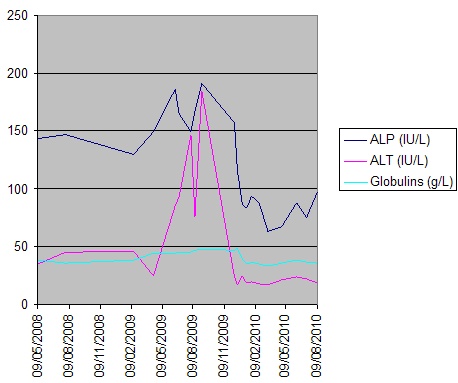

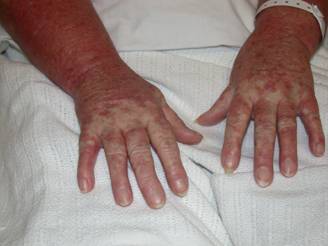
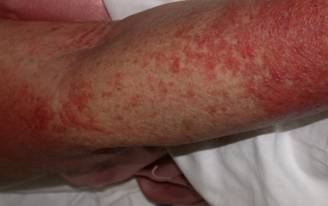
.jpg)

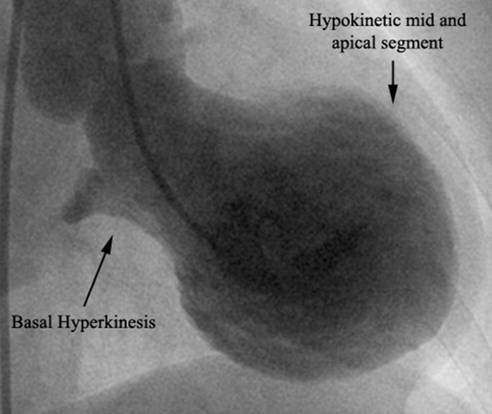
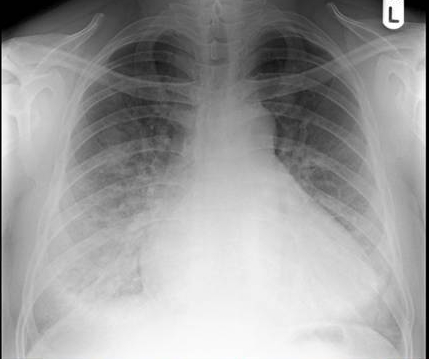
 Fig 1 (a): Pre-operative unenhanced CT scan which shows a large CSF density cystic lesion on the right side causing mass effect and midline shift to the left. There is no peri-lesional oedema. Fig 1 (b): Post-operative CT scan of the lesion shows a large void which can lead to dangerous collapse. Mild haematoma is also seen.
Fig 1 (a): Pre-operative unenhanced CT scan which shows a large CSF density cystic lesion on the right side causing mass effect and midline shift to the left. There is no peri-lesional oedema. Fig 1 (b): Post-operative CT scan of the lesion shows a large void which can lead to dangerous collapse. Mild haematoma is also seen.  Fig 2 (a): T1-weighted axial MRI of the brain demonstrates a cyst density similar to CSF. Fig 2 (b): T2-weighted MRI shows no ring enhancement or oedema. The periventricular hyperintensity of the left side is probably due to obstructive hydrocephalus.
Fig 2 (a): T1-weighted axial MRI of the brain demonstrates a cyst density similar to CSF. Fig 2 (b): T2-weighted MRI shows no ring enhancement or oedema. The periventricular hyperintensity of the left side is probably due to obstructive hydrocephalus. 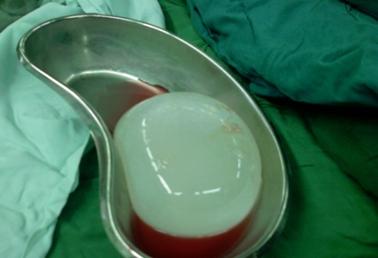 Fig 3: This shows the cyst removed in toto after operation. The cyst appears creamy and smooth. After summation of all the above data, the diagnosis of a hydatid cyst was made and a right frontotemporoparietal craniotomy was performed. A large cystic structure (14×14×12 cm) was delivered with utmost care to avoid rupture and spillage [fig 3]. A hydatid cyst was confirmed by pathology reports. A post-operative CT scan showed a large space without any residual matter [fig 1(b)]. Post-operatively, albendazole 15 mg/kg was started and continued for four weeks. The patient showed marked improvement in his neurological deficit and was discharged after one week with close follow-up. Discussion/Review Of Literature Life CycleHydatidosis is caused by Echinococcus granulosus, which occurs mainly in dogs. Humans who act as intermediate hosts get infected incidentally by ingesting eggs from the faeces of the infected animal. The eggs hatch inside the intestines and penetrate the walls, entering blood vessels and eventually reach the liver where they may form cysts or move on towards the lungs. Even after pulmonary filter, a few still make it to the systemic circulation and can lodge in almost any part of the body, including the brain, heart and bones.2,3,8,14,16,26 Brain hydatid cysts are relatively rare and only account for up to 2% of total cases.4,5,7 The actual percentage may be higher than what we have in literature, due to under-reporting. Brain hydatid cysts can be primary (single) or secondary (multiple).2,3,4,5,7 The latter are thought to arise from the multiple scolices released from the left side of the heart following cyst rupture in the heart2,3,5,27 or due to spontaneous, traumatic or surgical rupture of a solitary cranial cyst.3,5 Cysts mostly involve the territory of the middle cerebral artery4,7 but other regions like intraventricular, posterior fossa and the orbit have also been reported.15,17,18,28 The wall of the cyst consists of an inner endocyst (germinal layer) and outer ectocyst (laminated layer). The host reacts to the cyst forming a pericyst (fibrous capsule), which provides nutrients to the parasite. In the brain, due to minimal reaction, the pericyst is very thin. The endocyst produce scolices which bud into the cyst cavity and may sediment within the hydatid cavity, commonly known as hydatid sand.3,14,29,30 Presentation and DiagnosisMost hydatid cysts are acquired in childhood and are manifested during early adulthood.8,29 Cysts develop insidiously, usually being asymptomatic initially, and present with protean clinical and imaging features.3,5,6 In previous studies the most common presenting symptoms were headache and vomiting.4,5,7,14,15,28 Also in the literature, patients reported ataxia, diplopia, hemiparesis, abducens nerve palsy and even coma.5,7,15,28 Surprisingly, in the present study the patient did not have a headache and presented with parasthesia and numbness of the toes. Later he developed left sided weakness, convulsions and finally ataxia, which correlate with previous studies. Diagnosis of a hydatid cyst can sometimes be confused with other space occupying lesions of the brain, especially abscesses, neoplasms and arachnoid cysts.14,31 In this study the patient had bilateral frank papilloedema which is also mentioned in earlier reports.4,28 The Casoni and Weinberg tests, indirect haemagglutination, eosinophilia and ELISA are used in diagnosing hydatid cysts, but as brain tissue evokes minimal response many results tend to be false negatives.2,5,8,25 In our case also, serology for hydatid cyst was negative. CT scan and MRI are used frequently in diagnosing the cystic lesions.3,8,14,23,32,33 However, MRI is considered superior in demonstrating the cyst rim.5,8,11,21,32,34 On CT scan, a solitary cyst appears as well-defined, spherical, smooth, thin-walled and homogeneous, with an inner density similar to CSF, and non-enhancing walls.11,29,32The wall may appear iso-dense to hyper-dense on CT scan3,8, and rarely, may become calcified.11,29,32 There is usually no surrounding brain parenchymal oedema, which if exists along with ring enhancement, indicates inflammation and infection. 7,11,32,33,34,35 Ring enhancement and peri-lesional oedema differentiates brain abscesses and cystic neoplasms from uncomplicated hydatid cysts.3,8 These findings can in fact sometimes cause dilemma and misdiagnosis and lead to catastrophic events.14 The cyst shows low signal intensity on T1-weighted, and high signal intensity on T2-weighted MRI.2 MRI may also show peri-lesional oedema not seen on regular CT scan imaging.7 MRI may prove superior in determining exact cyst location, presence of super-added infections and cystic contents, and also in surgical planning and ruling out other diagnostic possibilities.14,33 We strongly recommend MRI for better evaluation of cystic brain lesions. Spontaneous cystic rupture can lead to different appearances depending on which layers have been obliterated, and produce some specific signs.3 When only the endocyst ruptures, cyst contents are held by the outer pericyst giving a peculiar water lily sign, which is pathognomic.3,8 TreatmentThough still in infancy, medical therapy for small or inoperable brain hydatid cysts has been promising. Albendazole alone or in combination with other compounds, such as praziquantel, has been reported with favourable results as an adjunct and, in certain circumstances, as the primary mode of treatment.2,36,37,38 It is reported that albendazole results in the disappearance of up to 48% of cysts and a substantial reduction in size of the cysts in another 28%.2 The duration of the treatment is four weeks or more, and recently many authors have favoured a prolonged therapy. The change in levels of cyst markers such as alanine, succinate, acetate and lactate, measured before and during treatment on Proton Magnetic Resonance Spectroscopy (MRS), correlate well with shrinkage and resolution of cyst findings on conventional MRI and help in evaluating the efficacy of chemotherapy.39 Cysts may drain into ventricles or rupture completely, causing spillage of contents into the subarachnoid space, leading to fatal anaphylactic shock, meningitis or local recurrence.3,5,22,25 Surgery is the mainstay for treating intracranial hydatid cysts and the aim is to excise the cysts entirely without rupture, which can otherwise lead to catastrophic events as described earlier 2,3,14,25. The Dowling-Orlando technique remains the preferred method, in which the cyst can be delivered by lowering the head of the operating table and instilling warm saline between the cyst and the surrounding brain.40 Even minimal spillage can cause deleterious effects (1 ml of hydatid sand contains 400,000 scolices).14 The thin cyst wall, periventricular location and micro-adhesions to the parenchyma are the main problems encountered during the surgical procedure.1,22 The large cavity remaining after the cystic removal can lead to many serious complications, such as cortical collapse, hyperpyrexia, brain oedema and cardio-respiratory failure.5 Recurrence remains a major concern, which is managed by both antihelminthic chemotherapy and surgery. In a study conducted by Ciurea et al, 25% of the patients had recurrence, which highlights the need for long term follow up.23 In the present study, due to the huge size of the cyst and progressive neurological deficit, it was not wise to completely rely on medical therapy. Surgery was performed and post-operatively albendazole was started as an adjunct. We recommend that for treating brain hydatid cyst, the size of the cyst, multiplicity, location and neurological deficit must all be taken into consideration.
Fig 3: This shows the cyst removed in toto after operation. The cyst appears creamy and smooth. After summation of all the above data, the diagnosis of a hydatid cyst was made and a right frontotemporoparietal craniotomy was performed. A large cystic structure (14×14×12 cm) was delivered with utmost care to avoid rupture and spillage [fig 3]. A hydatid cyst was confirmed by pathology reports. A post-operative CT scan showed a large space without any residual matter [fig 1(b)]. Post-operatively, albendazole 15 mg/kg was started and continued for four weeks. The patient showed marked improvement in his neurological deficit and was discharged after one week with close follow-up. Discussion/Review Of Literature Life CycleHydatidosis is caused by Echinococcus granulosus, which occurs mainly in dogs. Humans who act as intermediate hosts get infected incidentally by ingesting eggs from the faeces of the infected animal. The eggs hatch inside the intestines and penetrate the walls, entering blood vessels and eventually reach the liver where they may form cysts or move on towards the lungs. Even after pulmonary filter, a few still make it to the systemic circulation and can lodge in almost any part of the body, including the brain, heart and bones.2,3,8,14,16,26 Brain hydatid cysts are relatively rare and only account for up to 2% of total cases.4,5,7 The actual percentage may be higher than what we have in literature, due to under-reporting. Brain hydatid cysts can be primary (single) or secondary (multiple).2,3,4,5,7 The latter are thought to arise from the multiple scolices released from the left side of the heart following cyst rupture in the heart2,3,5,27 or due to spontaneous, traumatic or surgical rupture of a solitary cranial cyst.3,5 Cysts mostly involve the territory of the middle cerebral artery4,7 but other regions like intraventricular, posterior fossa and the orbit have also been reported.15,17,18,28 The wall of the cyst consists of an inner endocyst (germinal layer) and outer ectocyst (laminated layer). The host reacts to the cyst forming a pericyst (fibrous capsule), which provides nutrients to the parasite. In the brain, due to minimal reaction, the pericyst is very thin. The endocyst produce scolices which bud into the cyst cavity and may sediment within the hydatid cavity, commonly known as hydatid sand.3,14,29,30 Presentation and DiagnosisMost hydatid cysts are acquired in childhood and are manifested during early adulthood.8,29 Cysts develop insidiously, usually being asymptomatic initially, and present with protean clinical and imaging features.3,5,6 In previous studies the most common presenting symptoms were headache and vomiting.4,5,7,14,15,28 Also in the literature, patients reported ataxia, diplopia, hemiparesis, abducens nerve palsy and even coma.5,7,15,28 Surprisingly, in the present study the patient did not have a headache and presented with parasthesia and numbness of the toes. Later he developed left sided weakness, convulsions and finally ataxia, which correlate with previous studies. Diagnosis of a hydatid cyst can sometimes be confused with other space occupying lesions of the brain, especially abscesses, neoplasms and arachnoid cysts.14,31 In this study the patient had bilateral frank papilloedema which is also mentioned in earlier reports.4,28 The Casoni and Weinberg tests, indirect haemagglutination, eosinophilia and ELISA are used in diagnosing hydatid cysts, but as brain tissue evokes minimal response many results tend to be false negatives.2,5,8,25 In our case also, serology for hydatid cyst was negative. CT scan and MRI are used frequently in diagnosing the cystic lesions.3,8,14,23,32,33 However, MRI is considered superior in demonstrating the cyst rim.5,8,11,21,32,34 On CT scan, a solitary cyst appears as well-defined, spherical, smooth, thin-walled and homogeneous, with an inner density similar to CSF, and non-enhancing walls.11,29,32The wall may appear iso-dense to hyper-dense on CT scan3,8, and rarely, may become calcified.11,29,32 There is usually no surrounding brain parenchymal oedema, which if exists along with ring enhancement, indicates inflammation and infection. 7,11,32,33,34,35 Ring enhancement and peri-lesional oedema differentiates brain abscesses and cystic neoplasms from uncomplicated hydatid cysts.3,8 These findings can in fact sometimes cause dilemma and misdiagnosis and lead to catastrophic events.14 The cyst shows low signal intensity on T1-weighted, and high signal intensity on T2-weighted MRI.2 MRI may also show peri-lesional oedema not seen on regular CT scan imaging.7 MRI may prove superior in determining exact cyst location, presence of super-added infections and cystic contents, and also in surgical planning and ruling out other diagnostic possibilities.14,33 We strongly recommend MRI for better evaluation of cystic brain lesions. Spontaneous cystic rupture can lead to different appearances depending on which layers have been obliterated, and produce some specific signs.3 When only the endocyst ruptures, cyst contents are held by the outer pericyst giving a peculiar water lily sign, which is pathognomic.3,8 TreatmentThough still in infancy, medical therapy for small or inoperable brain hydatid cysts has been promising. Albendazole alone or in combination with other compounds, such as praziquantel, has been reported with favourable results as an adjunct and, in certain circumstances, as the primary mode of treatment.2,36,37,38 It is reported that albendazole results in the disappearance of up to 48% of cysts and a substantial reduction in size of the cysts in another 28%.2 The duration of the treatment is four weeks or more, and recently many authors have favoured a prolonged therapy. The change in levels of cyst markers such as alanine, succinate, acetate and lactate, measured before and during treatment on Proton Magnetic Resonance Spectroscopy (MRS), correlate well with shrinkage and resolution of cyst findings on conventional MRI and help in evaluating the efficacy of chemotherapy.39 Cysts may drain into ventricles or rupture completely, causing spillage of contents into the subarachnoid space, leading to fatal anaphylactic shock, meningitis or local recurrence.3,5,22,25 Surgery is the mainstay for treating intracranial hydatid cysts and the aim is to excise the cysts entirely without rupture, which can otherwise lead to catastrophic events as described earlier 2,3,14,25. The Dowling-Orlando technique remains the preferred method, in which the cyst can be delivered by lowering the head of the operating table and instilling warm saline between the cyst and the surrounding brain.40 Even minimal spillage can cause deleterious effects (1 ml of hydatid sand contains 400,000 scolices).14 The thin cyst wall, periventricular location and micro-adhesions to the parenchyma are the main problems encountered during the surgical procedure.1,22 The large cavity remaining after the cystic removal can lead to many serious complications, such as cortical collapse, hyperpyrexia, brain oedema and cardio-respiratory failure.5 Recurrence remains a major concern, which is managed by both antihelminthic chemotherapy and surgery. In a study conducted by Ciurea et al, 25% of the patients had recurrence, which highlights the need for long term follow up.23 In the present study, due to the huge size of the cyst and progressive neurological deficit, it was not wise to completely rely on medical therapy. Surgery was performed and post-operatively albendazole was started as an adjunct. We recommend that for treating brain hydatid cyst, the size of the cyst, multiplicity, location and neurological deficit must all be taken into consideration. 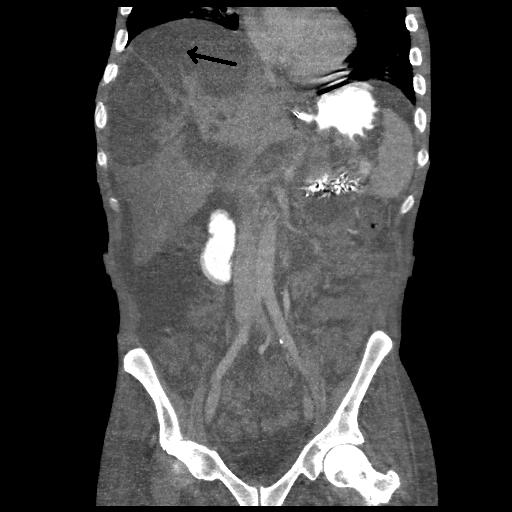
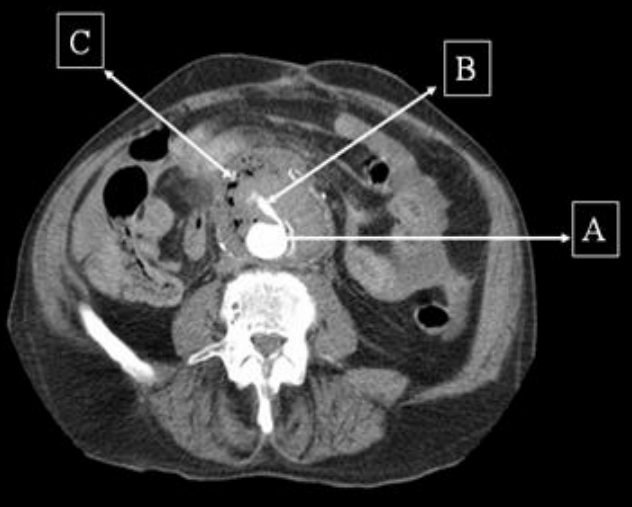

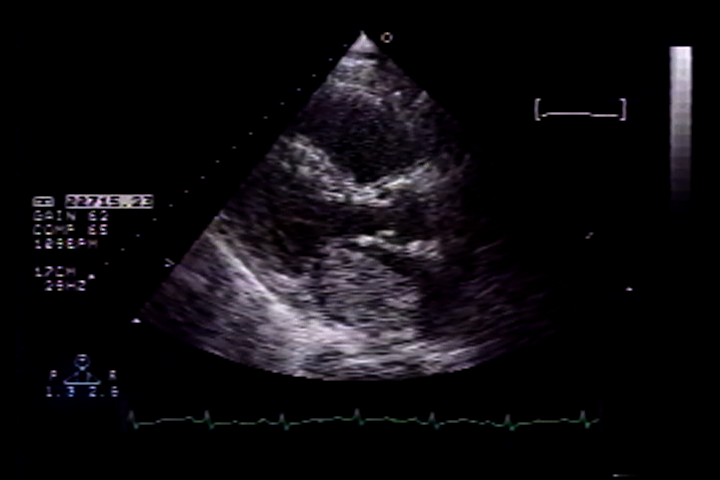

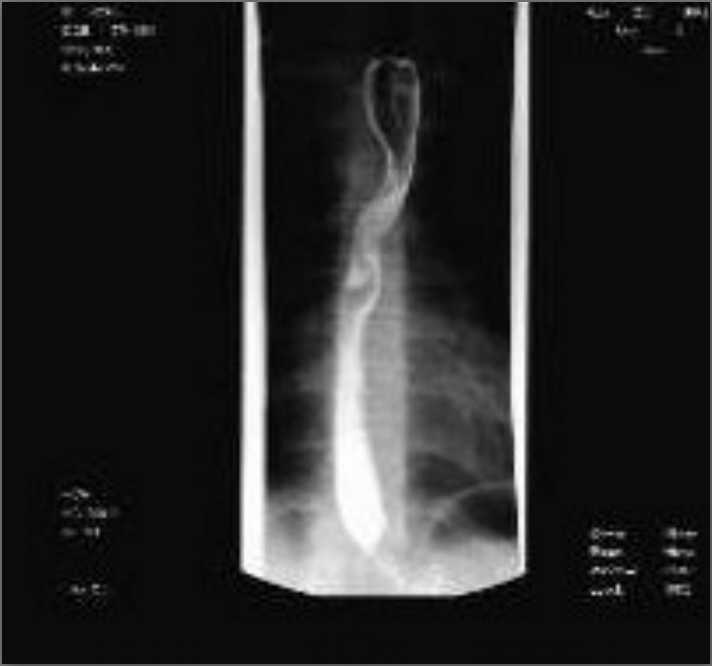
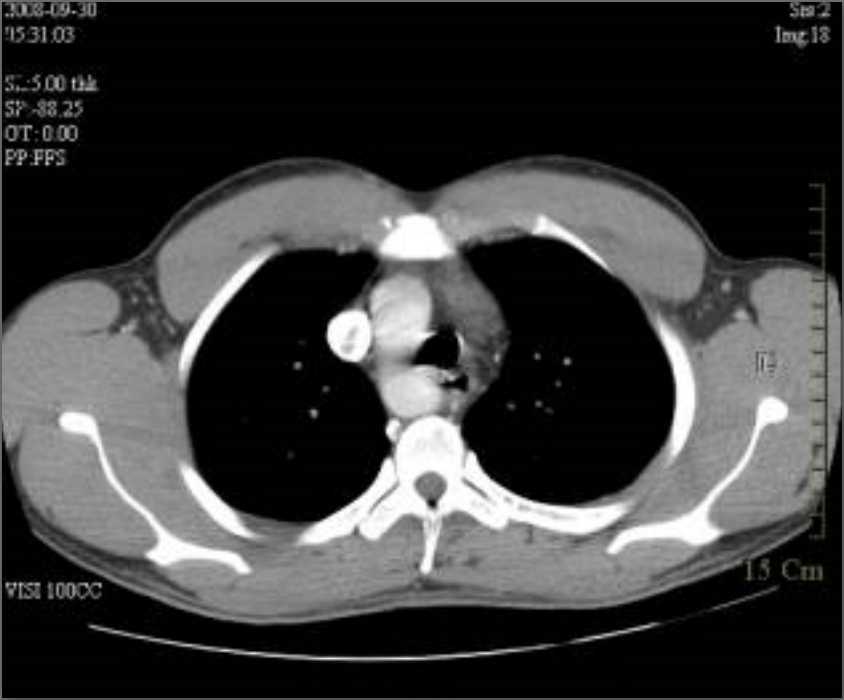

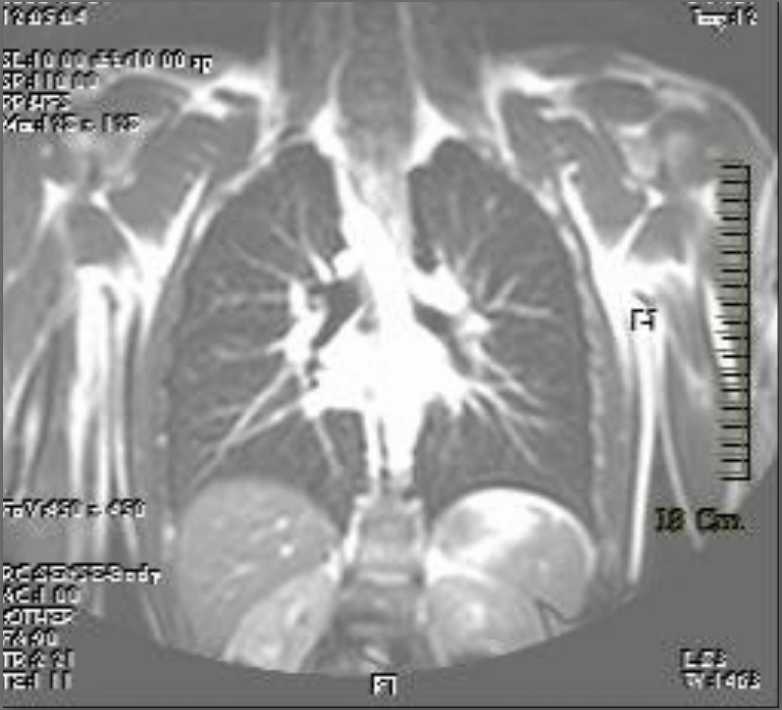
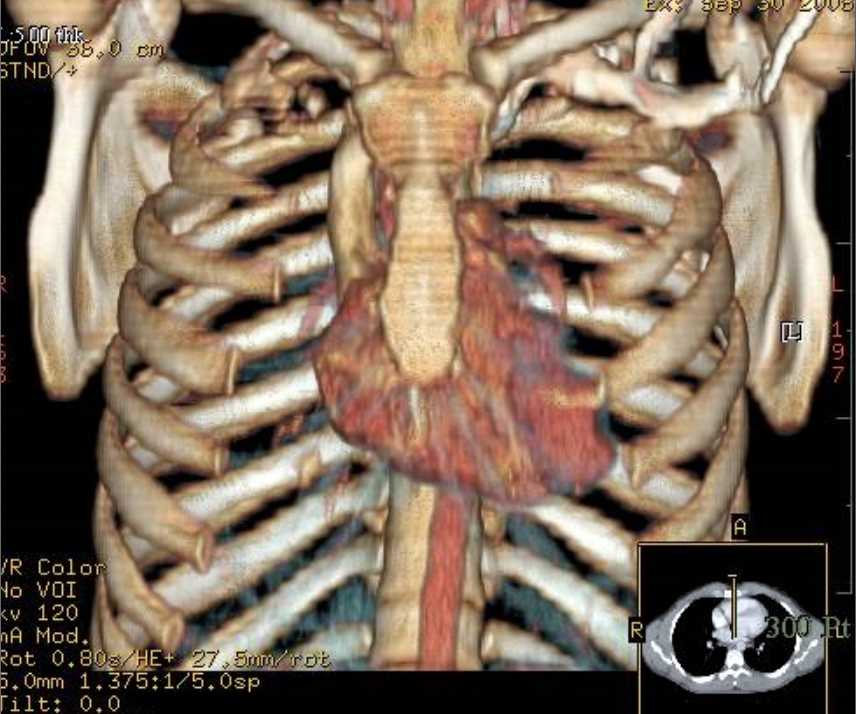
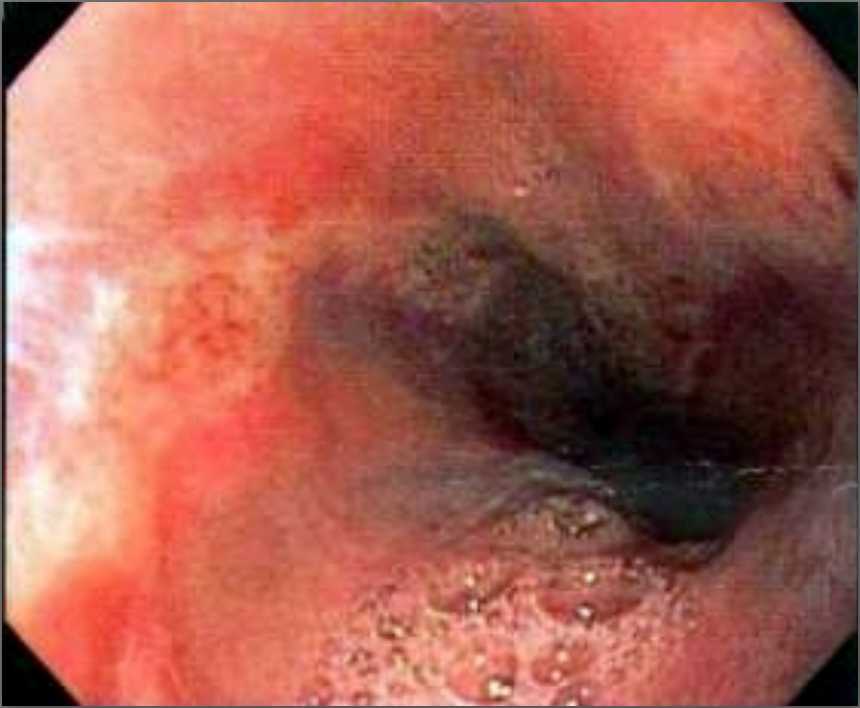
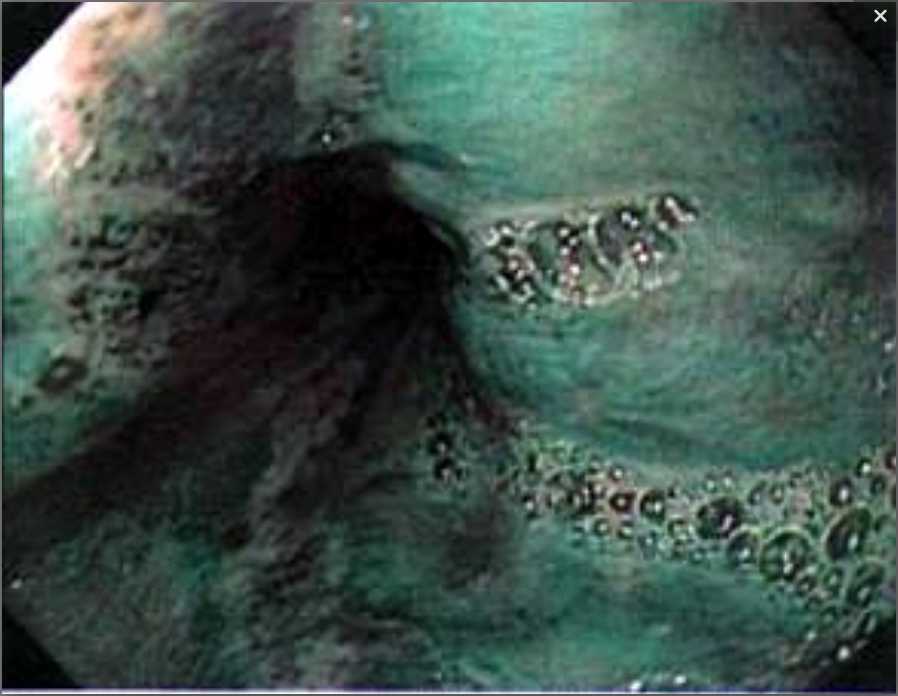
 Figure 1- Photograph showing gravid uterus lying in the incisional hernia sac The overlying skin was necrosed with evidence of ulceration and the presence of engorged veins. The fetus was lying in the herniated gravid uterus outside the abdominal cavity. Routine investigations were within normal limits. Ultrasound examination showed the uterus herniated in the incisional hernia of the anterior abdominal wall with the live fetus in cephalic presentation without any gross congenital malformation. The placenta was located in the upper uterine segment. She was kept in the hospital for bed rest with abdominal support. Emollients & antiseptic skin ointment were applied over the skin of the anterior abdominal wall. An elective caesarean section was planned for 37 weeks but she went into labour at 36 weeks. The abdomen was opened by elliptical incision. The uterus was visualized just beneath the skin and there was no evidence of the rectus sheath in the vicinity of the incision. A uterineincision was made over the previous caesarean scar and the baby was delivered with APGAR 7/10 at 1 minute and 9/10 at 5 minutes. The uterus was repaired in layers and a bilateral tubal ligation was done. Herniorraphy was performed in double buttress fashion. She was given a course of antibiotics. Her post operative period was uneventful and she went home with a healthy baby weighing 2.25 Kg. During her follow up visits she was found to be problem free. Discussion The remote complication of a caesarean section could be an incisional hernia due to defective abdominal wound healing and herniation of gravid uterus through the abdominal wall. This is a rare complication.1 The complications that have been reported in literature in association with this complication include strangulation, abortion, pre-term labour, accidental haemorrhage, intrauterine fetal death and rupture of the lower uterine segment.2 Excessive stretching of the skin may cause ulceration of the skin as in this present case due to friction between the hernia sac and other parts of the patient’s body. Caesarean section should be performed and herniorrhaphy can be performed during the caesarean section as in the present case.1 Herniorrhaphy can be performed during pregnancy if there is evidence of morbid incarceration or the skin is necrosed.3 However, herniorrhaphy can be postponed until delivery, as the enlarged uterus may interfere with healing of the repair.
Figure 1- Photograph showing gravid uterus lying in the incisional hernia sac The overlying skin was necrosed with evidence of ulceration and the presence of engorged veins. The fetus was lying in the herniated gravid uterus outside the abdominal cavity. Routine investigations were within normal limits. Ultrasound examination showed the uterus herniated in the incisional hernia of the anterior abdominal wall with the live fetus in cephalic presentation without any gross congenital malformation. The placenta was located in the upper uterine segment. She was kept in the hospital for bed rest with abdominal support. Emollients & antiseptic skin ointment were applied over the skin of the anterior abdominal wall. An elective caesarean section was planned for 37 weeks but she went into labour at 36 weeks. The abdomen was opened by elliptical incision. The uterus was visualized just beneath the skin and there was no evidence of the rectus sheath in the vicinity of the incision. A uterineincision was made over the previous caesarean scar and the baby was delivered with APGAR 7/10 at 1 minute and 9/10 at 5 minutes. The uterus was repaired in layers and a bilateral tubal ligation was done. Herniorraphy was performed in double buttress fashion. She was given a course of antibiotics. Her post operative period was uneventful and she went home with a healthy baby weighing 2.25 Kg. During her follow up visits she was found to be problem free. Discussion The remote complication of a caesarean section could be an incisional hernia due to defective abdominal wound healing and herniation of gravid uterus through the abdominal wall. This is a rare complication.1 The complications that have been reported in literature in association with this complication include strangulation, abortion, pre-term labour, accidental haemorrhage, intrauterine fetal death and rupture of the lower uterine segment.2 Excessive stretching of the skin may cause ulceration of the skin as in this present case due to friction between the hernia sac and other parts of the patient’s body. Caesarean section should be performed and herniorrhaphy can be performed during the caesarean section as in the present case.1 Herniorrhaphy can be performed during pregnancy if there is evidence of morbid incarceration or the skin is necrosed.3 However, herniorrhaphy can be postponed until delivery, as the enlarged uterus may interfere with healing of the repair. Image 1: Initial chest x-ray revealing bronchogenic cyst in the posterior right middle lobe (10x10x8 cm3) Computerized tomography (CT) scan showed a large cystic lesion arising entirely within the right lower lobe and extending the width of the hemithorax. (Image 2)
Image 1: Initial chest x-ray revealing bronchogenic cyst in the posterior right middle lobe (10x10x8 cm3) Computerized tomography (CT) scan showed a large cystic lesion arising entirely within the right lower lobe and extending the width of the hemithorax. (Image 2) 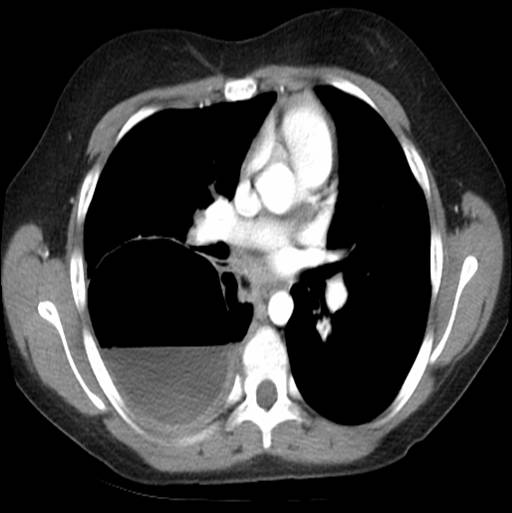 Image 2: Chest CT shows bronchogenic cyst extending the entire width of the right hemithorax and approximately 50% full of fluid. There was an air-fluid level occupying ~50% of the cavity. She was diagnosed with a multilocular bronchogenic cyst. She was briefly hospitalized and discharged on azithromycin with plans to resect the cyst in one month. Severe cough, fever, and chills prompted readmission after 3 weeks of antibiotic therapy. CXR and CT showed cyst enlargement (16x9x11 cm3) with over 95% fluid. (Images 3 and 4)
Image 2: Chest CT shows bronchogenic cyst extending the entire width of the right hemithorax and approximately 50% full of fluid. There was an air-fluid level occupying ~50% of the cavity. She was diagnosed with a multilocular bronchogenic cyst. She was briefly hospitalized and discharged on azithromycin with plans to resect the cyst in one month. Severe cough, fever, and chills prompted readmission after 3 weeks of antibiotic therapy. CXR and CT showed cyst enlargement (16x9x11 cm3) with over 95% fluid. (Images 3 and 4) 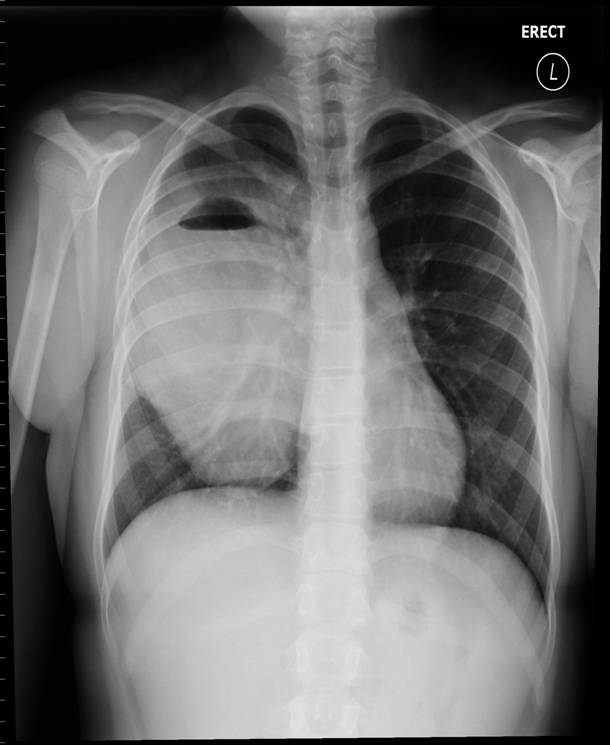 Image 3: Substantial bronchogenic cyst (16x9x11 cm3), over 95% full of fluid.
Image 3: Substantial bronchogenic cyst (16x9x11 cm3), over 95% full of fluid. 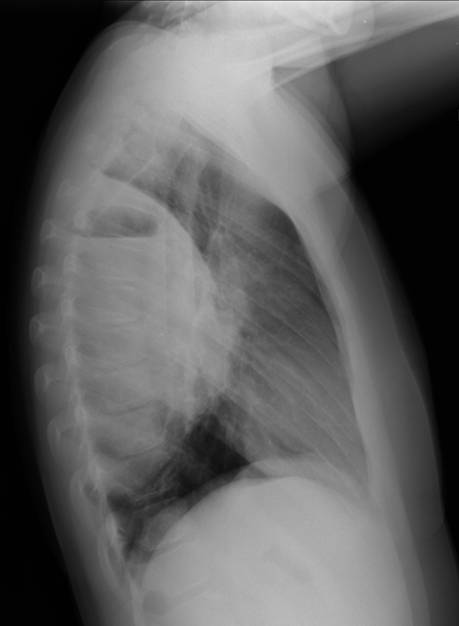 Image 4: Lateral chest x-ray revealed opacification along superior margin of cyst. She was started on ampicillin/sulbactam. Percutaneous drain placement yielded a large volume of turbid fluid. Aerobe, anaerobe and fungal studies of the fluid were negative. Resection was postponed due to significant inflammation surrounding the cyst cavity. She was discharged on a seven day course of amoxicillin/clavulanate. Following six weeks of cyst drainage, a thoracoscopic right lower lobectomy was performed. Extensive inflammation and induration made dissection of the lower lobe and pulmonary vessels challenging. Fibrinoid adhesions extended to the pleural surface. Operative time was 418 minutes. Surgical pathology showed diffuse necrotizing granulomatous inflammation with acid-fast bacilli and multiple nodules up to 3.3 cm in diameter. Ninety-five percent of the pleural surface had nodular involvement. (Image 5) Areas of non-indurated lung also showed small nodules with a miliary appearance. Inflammation was present at the bronchovascular margins, hilar nodes, and distal lung.
Image 4: Lateral chest x-ray revealed opacification along superior margin of cyst. She was started on ampicillin/sulbactam. Percutaneous drain placement yielded a large volume of turbid fluid. Aerobe, anaerobe and fungal studies of the fluid were negative. Resection was postponed due to significant inflammation surrounding the cyst cavity. She was discharged on a seven day course of amoxicillin/clavulanate. Following six weeks of cyst drainage, a thoracoscopic right lower lobectomy was performed. Extensive inflammation and induration made dissection of the lower lobe and pulmonary vessels challenging. Fibrinoid adhesions extended to the pleural surface. Operative time was 418 minutes. Surgical pathology showed diffuse necrotizing granulomatous inflammation with acid-fast bacilli and multiple nodules up to 3.3 cm in diameter. Ninety-five percent of the pleural surface had nodular involvement. (Image 5) Areas of non-indurated lung also showed small nodules with a miliary appearance. Inflammation was present at the bronchovascular margins, hilar nodes, and distal lung.  Image 5: Gross specimen of right lower lobe: Approximately half of the lobe was indurated and 95% of surfaces showed nodular involvement. Sectioning through indurated region revealed diffuse nodules up to 3.3 cm. Nonindurated lung showed small nodules with miliary appearance. The patient had no history of tuberculosis exposure, foreign travel or immunodeficiency. There was no family history of tuberculosis or respiratory disease. Based on the acid-fast bacilli identified on pathology stain, fluid drained from her chest tube was sent for acid-fast bacilli culture and smear. Mycobacterium was not isolated. It was determined that the source of the atypical mycobacterial infection was likely colonizing mycobacteria from her oropharynx that became entrapped in the cyst. A six-week course of clarithromycin, rifampin, and ethambutol was prescribed to treat any remaining organisms. At two-month follow-up, she had minimal pulmonary symptoms and inflammatory markers were improved. Erythrocyte sedimentation rate (normal: 0-15) and C-reactive protein level (normal: 0-10) decreased from 88 and 173 during her hospitalization, to 10 and 3.6, respectively. At four-month follow-up, she had resumed competitive sports and had no evidence of ongoing infection. Discussion: This case highlights a unique presentation of infected bronchogenic cyst after substantial cyst growth. Unusual aspects include the late onset of symptoms, multilocular intraparenchymal cyst appearance, turbid drainage, extensive nodularity, necrotizing granulomatous inflammation, and atypical Mycobacterium infection. Although comorbid infection is not uncommon, causative organisms are typically Haemophilus influenzae1,2 and Streptococcus pneumoniae.3 Cases of Streptococcus pyogenes,4 Escherichia coli,5and Salmonella enteritidis6have been reported. However, only four cases of bronchogenic cyst with Mycobacterium infection have been documented.7,8,9 Three of the Mycobacterium-infected cases are adult patients. Lin et. al reported a 39-year-old female with bronchogenic cyst complicated by Mycobacterium avium infection.7 The organism was identified by genetic sequencing of biopsied lung tissue. Sputum acid-fast stain and mycobacterial cultures were negative. Liman et al. reported two adult cases: a 20 year-old male with Mycobacterium identified in a right lower lobe specimen but with negative sputum culture, and a 32 year-old female with Mycobacterium isolated in a sputum culture but a negative microscopic exam and cyst fluid culture.8 The only documented paediatric case, a 9 year-old female with a 6 cm right lower lobe bronchogenic cyst, was reported by Houser et al.9 She underwent lobectomy; Kinyoun stain of the cyst specimen showed Mycobacterium. Sputum culture and acid-fast bacilli stain were negative. Tuberculin skin test was positive. Comorbid infection with Mycobacterium tuberculosis was suggested, but they were unable to isolate an organism. Treatment consisted of four months of rifampin and two years of isoniazid with pyridoxine. This is the first documented paediatric case of bronchogenic cyst infected with atypical Mycobacterium. Her presentation is noteworthy, given the substantially greater size of the cyst (16x9x11 cm), extensive pathologic findings, and success with a different antibiotic regimen.
Image 5: Gross specimen of right lower lobe: Approximately half of the lobe was indurated and 95% of surfaces showed nodular involvement. Sectioning through indurated region revealed diffuse nodules up to 3.3 cm. Nonindurated lung showed small nodules with miliary appearance. The patient had no history of tuberculosis exposure, foreign travel or immunodeficiency. There was no family history of tuberculosis or respiratory disease. Based on the acid-fast bacilli identified on pathology stain, fluid drained from her chest tube was sent for acid-fast bacilli culture and smear. Mycobacterium was not isolated. It was determined that the source of the atypical mycobacterial infection was likely colonizing mycobacteria from her oropharynx that became entrapped in the cyst. A six-week course of clarithromycin, rifampin, and ethambutol was prescribed to treat any remaining organisms. At two-month follow-up, she had minimal pulmonary symptoms and inflammatory markers were improved. Erythrocyte sedimentation rate (normal: 0-15) and C-reactive protein level (normal: 0-10) decreased from 88 and 173 during her hospitalization, to 10 and 3.6, respectively. At four-month follow-up, she had resumed competitive sports and had no evidence of ongoing infection. Discussion: This case highlights a unique presentation of infected bronchogenic cyst after substantial cyst growth. Unusual aspects include the late onset of symptoms, multilocular intraparenchymal cyst appearance, turbid drainage, extensive nodularity, necrotizing granulomatous inflammation, and atypical Mycobacterium infection. Although comorbid infection is not uncommon, causative organisms are typically Haemophilus influenzae1,2 and Streptococcus pneumoniae.3 Cases of Streptococcus pyogenes,4 Escherichia coli,5and Salmonella enteritidis6have been reported. However, only four cases of bronchogenic cyst with Mycobacterium infection have been documented.7,8,9 Three of the Mycobacterium-infected cases are adult patients. Lin et. al reported a 39-year-old female with bronchogenic cyst complicated by Mycobacterium avium infection.7 The organism was identified by genetic sequencing of biopsied lung tissue. Sputum acid-fast stain and mycobacterial cultures were negative. Liman et al. reported two adult cases: a 20 year-old male with Mycobacterium identified in a right lower lobe specimen but with negative sputum culture, and a 32 year-old female with Mycobacterium isolated in a sputum culture but a negative microscopic exam and cyst fluid culture.8 The only documented paediatric case, a 9 year-old female with a 6 cm right lower lobe bronchogenic cyst, was reported by Houser et al.9 She underwent lobectomy; Kinyoun stain of the cyst specimen showed Mycobacterium. Sputum culture and acid-fast bacilli stain were negative. Tuberculin skin test was positive. Comorbid infection with Mycobacterium tuberculosis was suggested, but they were unable to isolate an organism. Treatment consisted of four months of rifampin and two years of isoniazid with pyridoxine. This is the first documented paediatric case of bronchogenic cyst infected with atypical Mycobacterium. Her presentation is noteworthy, given the substantially greater size of the cyst (16x9x11 cm), extensive pathologic findings, and success with a different antibiotic regimen.